5 AverageHeatmap
AverageHeatmap is used to plot averaged expression cross cluster cells.
5.1 load data
httest <- system.file("extdata", "htdata.RDS", package = "scRNAtoolVis")
pbmc <- readRDS(httest)
# load markergene
markergene <- system.file("extdata", "top5pbmc.markers.csv", package = "scRNAtoolVis")
markers <- read.table(markergene, sep = ',', header = TRUE)5.2 examples
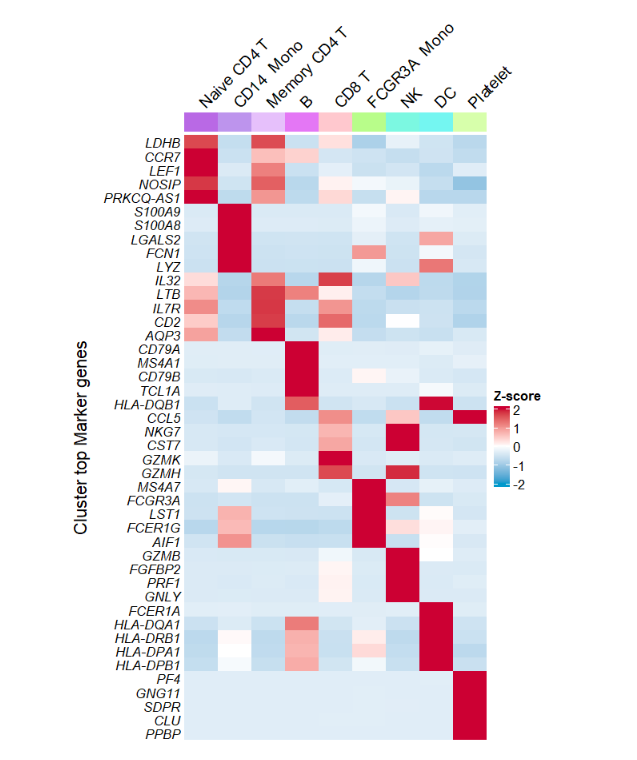
default plot:
# plot
AverageHeatmap(object = pbmc,
markerGene = markers$gene)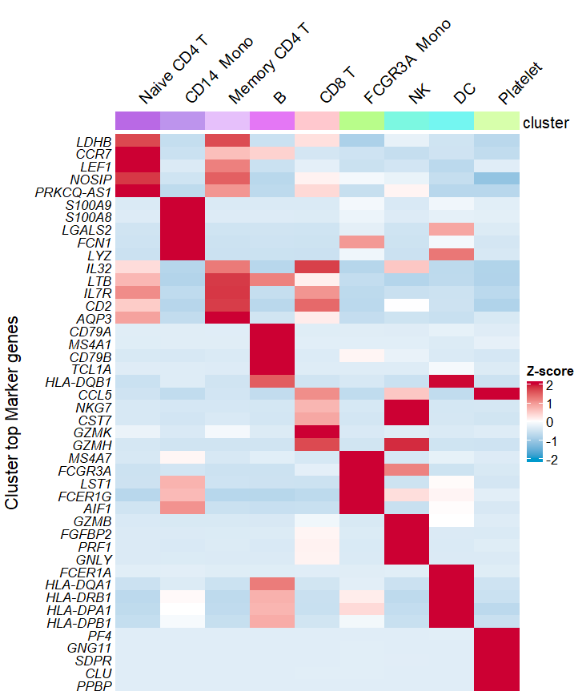 change color:
change color:
# change color
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
htCol = c("#339933", "#FFCC00", "#FF0033"))
Supporting with your own cluster colors by annoCol = TRUE and myanCol:
# change annotation color
library("scales")
library(ggsci)
mycol <- hue_pal()(9)
mycol1 <- pal_npg()(9)
# plot
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
annoCol = TRUE,
myanCol = mycol) +
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
annoCol = TRUE,
myanCol = mycol1)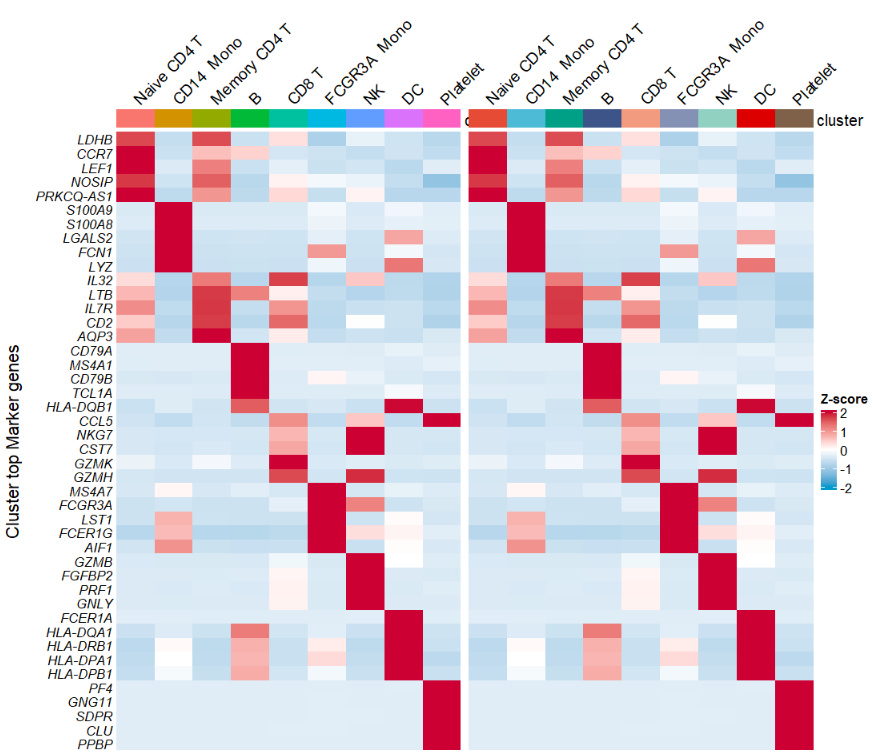
Remove rownames:
# remove rownames
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
showRowNames = F) Remove annotation name:
Remove annotation name:
# remove cluster anno name
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
clusterAnnoName = F)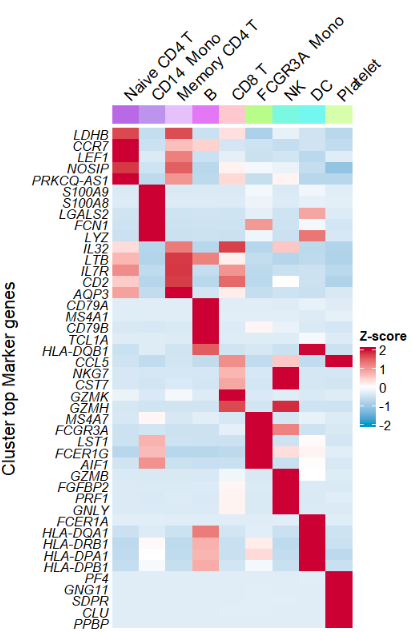 Mark some important genes:
Mark some important genes:
# mark some genes
# tartget gene
annoGene <- c("LDHB","CCR7","LEF1","NKG7","CST7",
"GZMK","HLA-DQA1","HLA-DRB1","HLA-DPA1")
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
clusterAnnoName = F,
showRowNames = F,
markGenes = annoGene)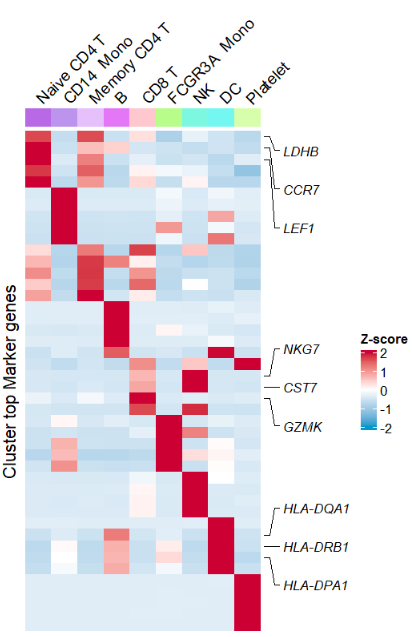
You can change heatmap width and height:
# change heatmap width and height
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
clusterAnnoName = F,
width = 8,height = 16)