Chapter 7 Hic heatmap visualization
The related Hic data for input we have discussed before. prepareHic function will be used in the data preparation step. Here we use the test data in plotgardenerData package as examples. Package plotgardener will outputs the following results:
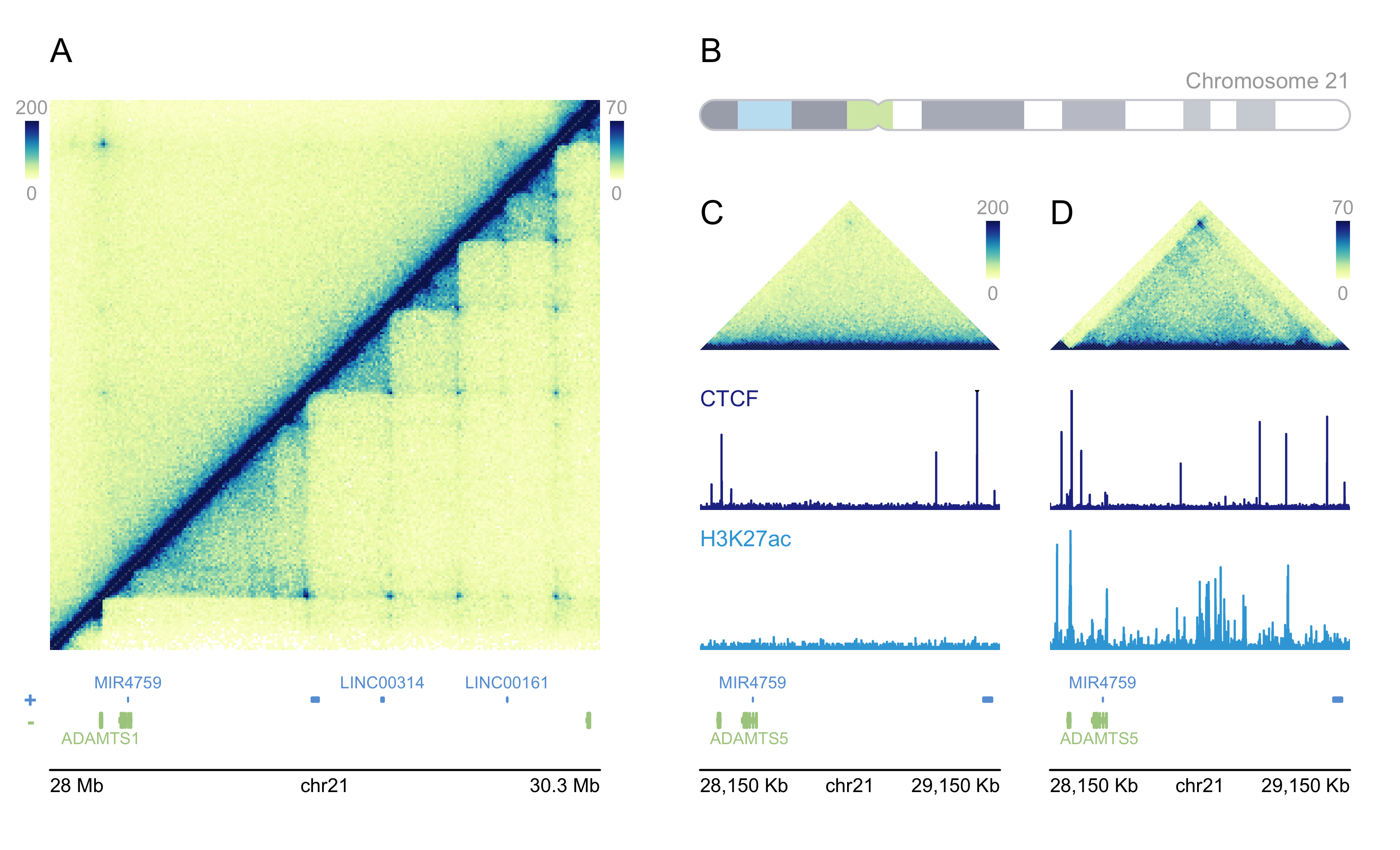
Let’s use trackVisProMax to finish it.
7.1 Data preperation
Load Hic data:
library(plotgardenerData)
library(plotgardener)
data("GM12878_HiC_10kb")
data("IMR90_HiC_10kb")
data("GM12878_ChIP_CTCF_signal")
data("IMR90_ChIP_CTCF_signal")
data("GM12878_ChIP_H3K27ac_signal")
data("IMR90_ChIP_H3K27ac_signal")
# prepare hic data
data = list(GM12878_HiC_10kb,IMR90_HiC_10kb)
file_name = c("GM12878_HiC","IMR90_HiC")
chrom = c("21","21")
resolution = c(10000,10000)
input_hic <- prepareHic(data = list(GM12878_HiC_10kb,IMR90_HiC_10kb),
file_name = c("GM12878_HiC","IMR90_HiC"),
chrom = c("21","21"),
resolution = c(10000,10000))Load bigwig data:
# prepare data
hic_bw <- rbind(GM12878_ChIP_CTCF_signal,IMR90_ChIP_CTCF_signal,
GM12878_ChIP_H3K27ac_signal,IMR90_ChIP_H3K27ac_signal)
hic_bw$fileName <- rep(c("CTCF_signal","IMR90_CTCF_signal",
"H3K27ac_signal","IMR90_H3K27ac_signal"),
c(nrow(GM12878_ChIP_CTCF_signal),nrow(IMR90_ChIP_CTCF_signal),
nrow(GM12878_ChIP_H3K27ac_signal),nrow(IMR90_ChIP_H3K27ac_signal)))
colnames(hic_bw)[1] <- "seqnames"
# gtf
gtf <- rtracklayer::import.gff("test-bw2/hg19.ncbiRefSeq.gtf.gz",format = "gtf") %>%
data.frame() 7.2 Basic plot
Let’s see the bigwig track data first:
# plot
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("CTCF_signal","H3K27ac_signal")),
Input_gtf = gtf,
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj))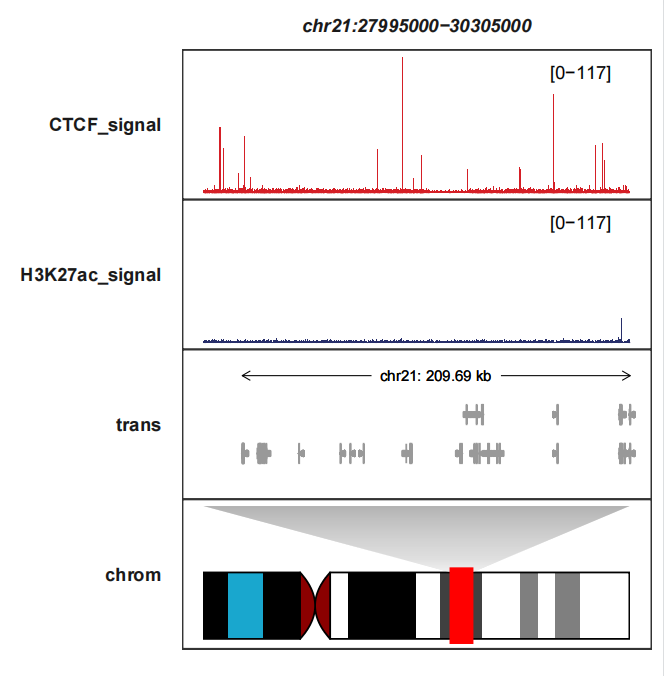
Adding heatmap track:
# plot
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("CTCF_signal","H3K27ac_signal")),
Input_gtf = gtf,
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj))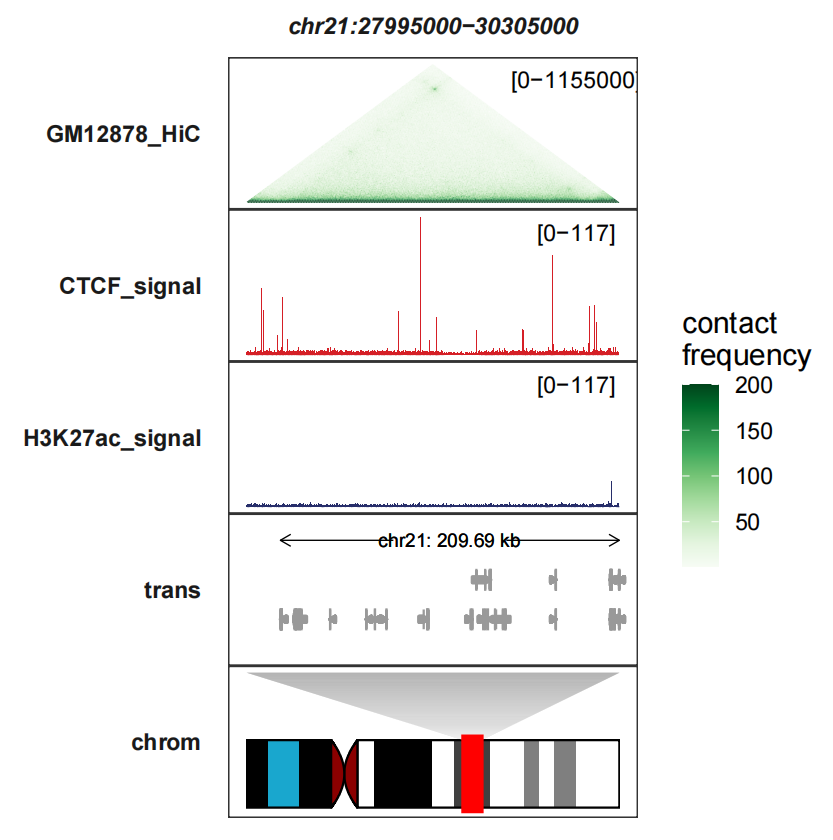
Let’s zoom the heatmap and see details:

Removing the legend:
# plot
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("CTCF_signal","H3K27ac_signal")),
Input_hic = input_hic %>% filter(fileName == "GM12878_HiC"),
Input_gtf = gtf,
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
fixed_column_range = F,
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj),
sample_order = c("GM12878_HiC","CTCF_signal","H3K27ac_signal"),
signal_range = list("chr21:27995000-30305000" = c(CTCF_signal = 117,H3K27ac_signal = 117)),
signal_layer_heatmap_params = list(show.legend = F))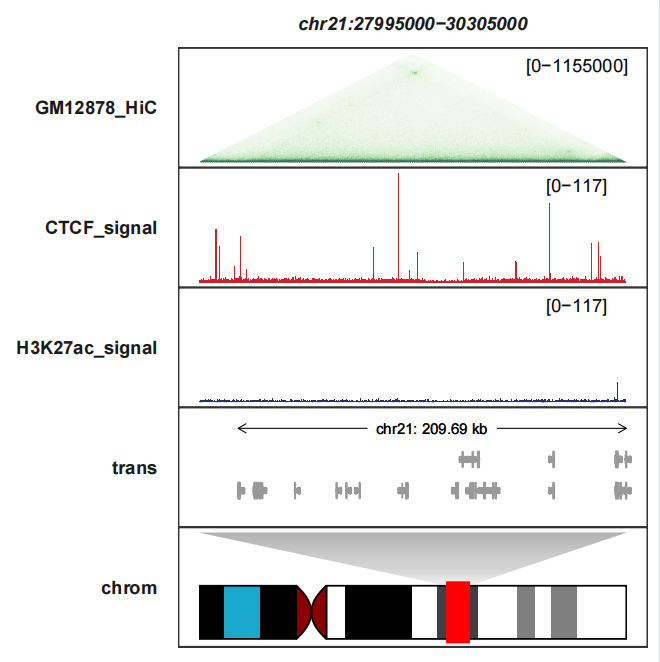
Seeing another example track on the right:
# add contact heatmap
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("IMR90_CTCF_signal","IMR90_H3K27ac_signal")),
Input_gtf = gtf,
Input_hic = input_hic %>% filter(fileName == "IMR90_HiC"),
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
fixed_column_range = F,
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj),
sample_order = c("IMR90_HiC","IMR90_CTCF_signal","IMR90_H3K27ac_signal"),
signal_range = list("chr21:27995000-30305000" =
c(IMR90_CTCF_signal = 77,IMR90_H3K27ac_signal = 77)))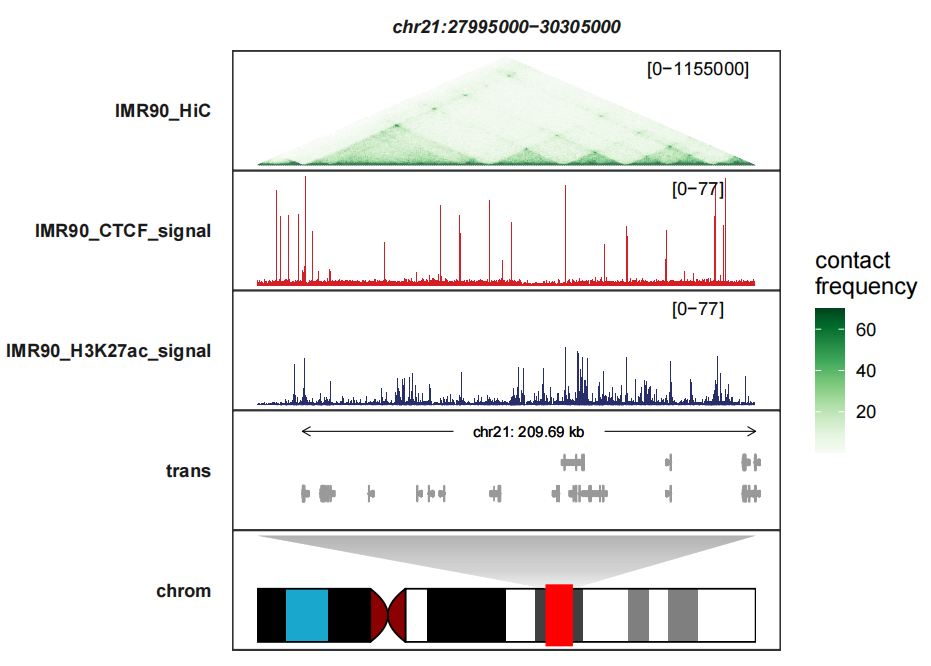
heatmap_fill_col controls the heatmap colors, here we use same colors corresponding to the example:
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("IMR90_CTCF_signal","IMR90_H3K27ac_signal")),
Input_gtf = gtf,
Input_hic = input_hic %>% filter(fileName == "IMR90_HiC"),
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
fixed_column_range = F,
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj),
sample_order = c("IMR90_HiC","IMR90_CTCF_signal","IMR90_H3K27ac_signal"),
signal_range = list("chr21:27995000-30305000" =
c(IMR90_CTCF_signal = 77,IMR90_H3K27ac_signal = 77)),
heatmap_fill_col = RColorBrewer::brewer.pal(n = 9, "YlGnBu"))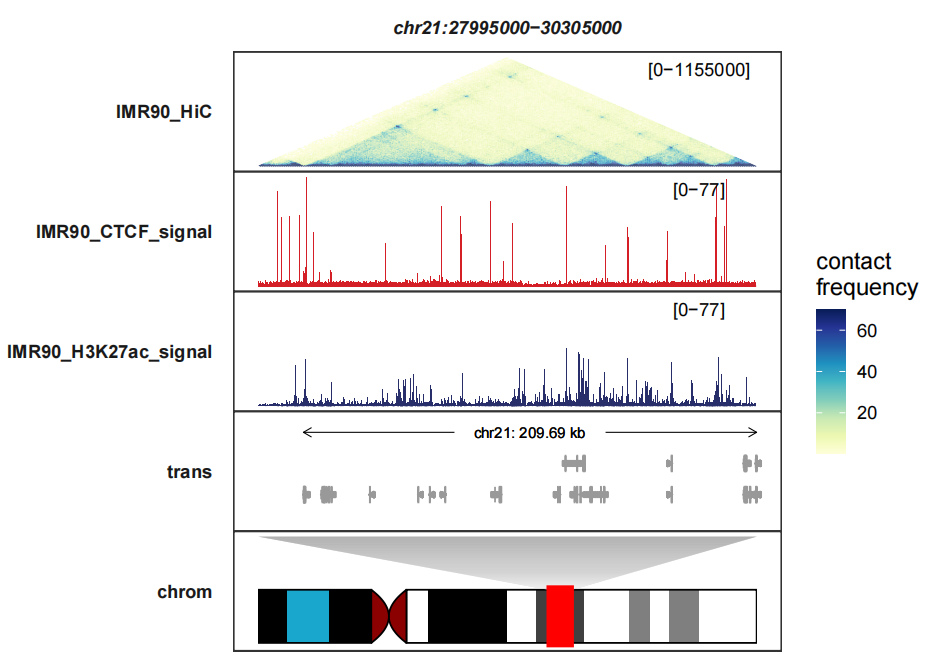
Reverting the heatmap:
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("IMR90_CTCF_signal","IMR90_H3K27ac_signal")),
Input_gtf = gtf,
Input_hic = input_hic %>% filter(fileName == "IMR90_HiC"),
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
fixed_column_range = F,
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj),
sample_order = c("IMR90_CTCF_signal","IMR90_HiC","IMR90_H3K27ac_signal"),
signal_range = list("chr21:27995000-30305000" =
c(IMR90_CTCF_signal = 77,IMR90_H3K27ac_signal = 77)),
reverse_y_vars = c("IMR90_HiC"),
heatmap_fill_col = RColorBrewer::brewer.pal(n = 9, "RdYlBu"))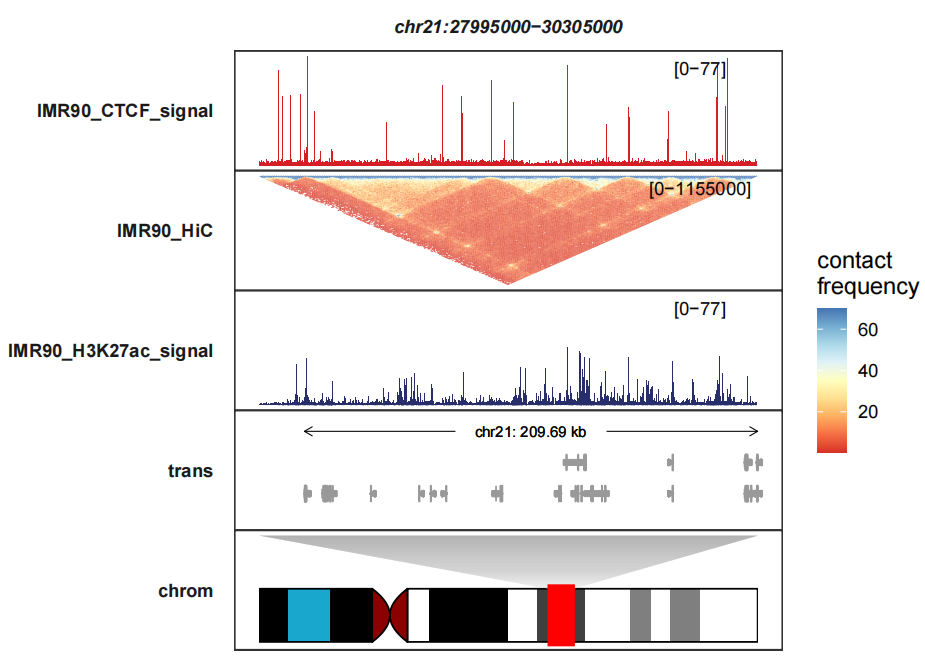
You will see some uncomplete graphic elements beside the heatmap when you change into other genomic regions:
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("IMR90_CTCF_signal","IMR90_H3K27ac_signal")),
Input_gtf = gtf,
Input_hic = input_hic %>% filter(fileName == "IMR90_HiC"),
query_region = list(query_chr = "chr21",
query_start = 29500000,
query_end = 30000000),
fixed_column_range = F,
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj),
sample_order = c("IMR90_HiC","IMR90_CTCF_signal","IMR90_H3K27ac_signal"),
signal_range = list("chr21:27995000-30305000" =
c(IMR90_HiC = 400000,IMR90_CTCF_signal = 77,
IMR90_H3K27ac_signal = 77)),
heatmap_fill_col = RColorBrewer::brewer.pal(n = 9, "YlGnBu"))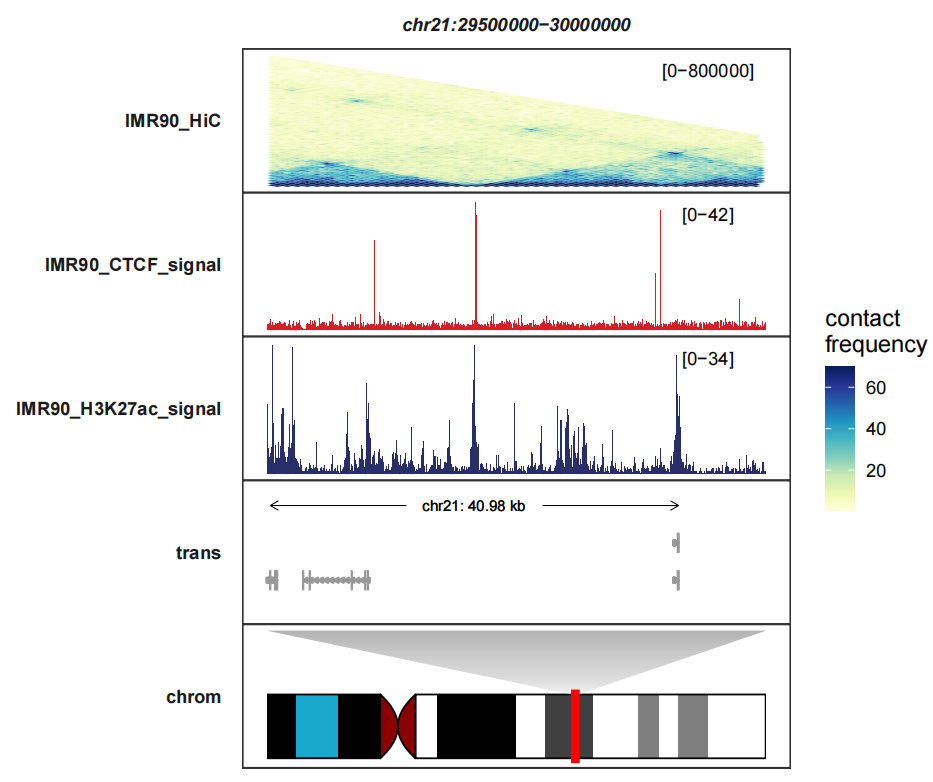
Details beside the heatmap. This is because some polygon positions were filtered which are outside the specified genomic regions:

You can use xlimit_range parameter to zoom a region:
trackVisProMax(Input_bw = hic_bw %>%
filter(fileName %in% c("IMR90_CTCF_signal","IMR90_H3K27ac_signal")),
Input_gtf = gtf,
Input_hic = input_hic %>% filter(fileName == "IMR90_HiC"),
query_region = list(query_chr = "chr21",
query_start = 27995000,
query_end = 30305000),
fixed_column_range = F,
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj),
sample_order = c("IMR90_HiC","IMR90_CTCF_signal","IMR90_H3K27ac_signal"),
signal_range = list("chr21:27995000-30305000" =
c(IMR90_CTCF_signal = 77,
IMR90_H3K27ac_signal = 77)),
heatmap_fill_col = RColorBrewer::brewer.pal(n = 9, "YlGnBu"),
xlimit_range = c(29500000,30000000))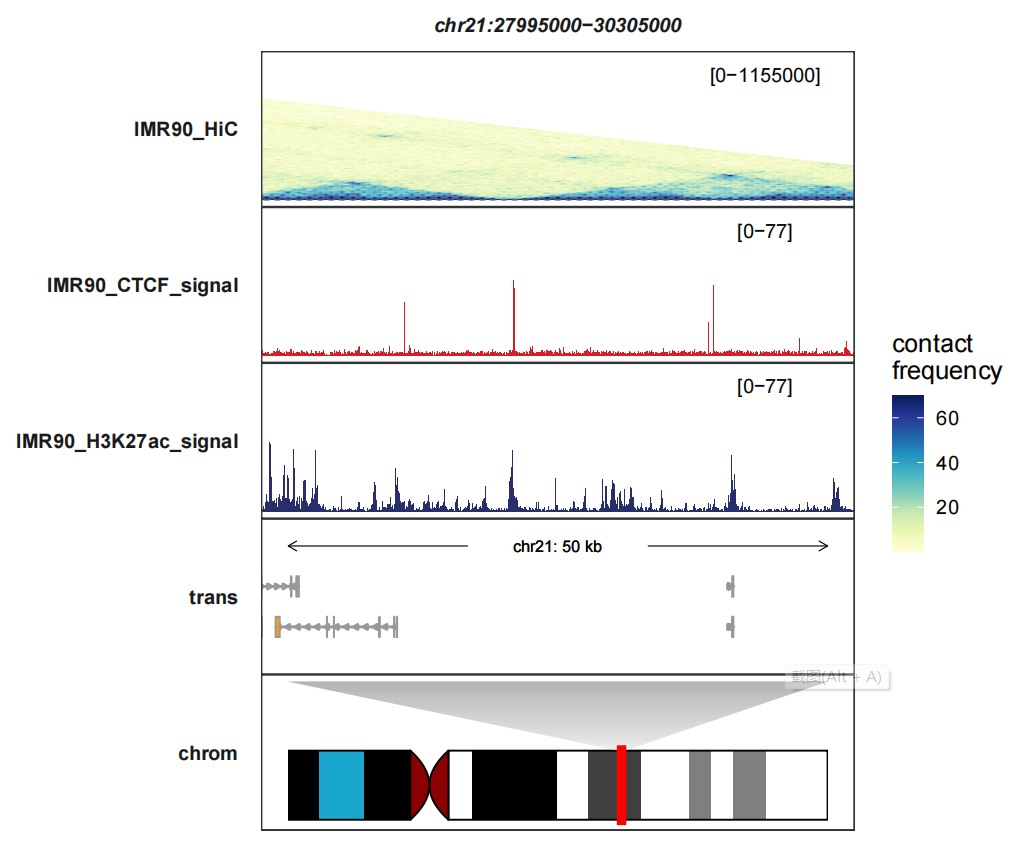
7.3 Real data practice
Note: Using chromstart and chromend to extarct data from a specified genomic region and setting a suitable resolution.
Here we do a practice with a public Hic data.
Test data can be fetched on GSE200160.
First extract heatmap data from .hic data from a specified genomic region:
# real data
hic_data <- list.files("test-bw2/",pattern = ".hic",full.names = T)
hic_data
# [1] "test-bw2/RPE-doxorubicin_02uM.hic" "test-bw2/RPE-doxorubicin_034uM.hic"
# [3] "test-bw2/RPE-ICRF193_5uM.hic"
hic_df <- prepareHic(hic_path = hic_data,
file_name = c("doxorubicin_02uM","doxorubicin_34uM",
"ICRF193_5uM"),
chrom = "1",assembly = "hg19",
chromstart = 29400000,chromend = 29500000,
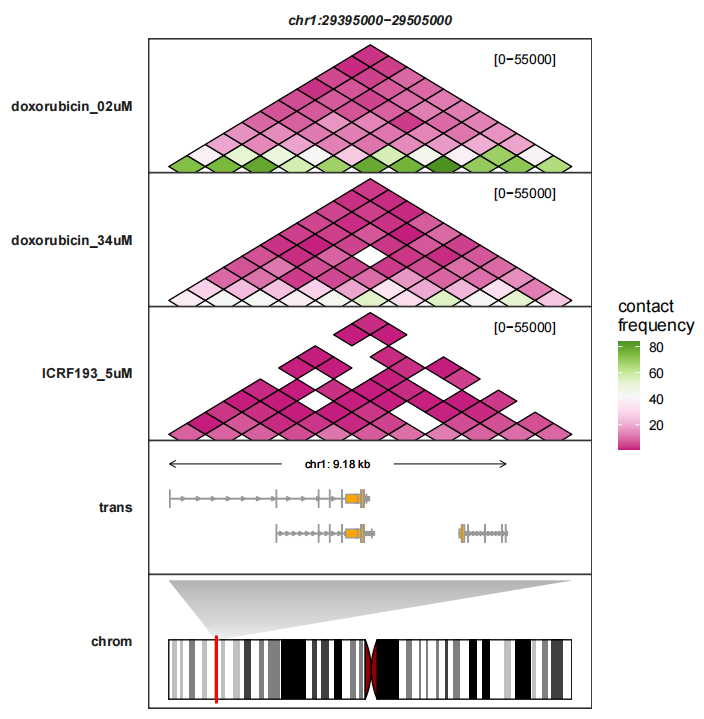
resolution = 10000)Now plot this region:
# plot
trackVisProMax(Input_gtf = gtf,
Input_hic = hic_df,
query_region = list(query_chr = "chr1",
query_start = min(hic_df$start),
query_end = max(hic_df$start)),
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj))
Add border colors and change heatmap fill colors:
# plot
trackVisProMax(Input_gtf = gtf,
Input_hic = hic_df,
query_region = list(query_chr = "chr1",
query_start = min(hic_df$start),
query_end = max(hic_df$start)),
draw_chromosome = T,
draw_chromosome_params = list(ideogram_obj = hg19_obj))