Chapter 9 Adding gene labels
Here shows an example to add gene names from gtf file on genome track. You only need to extract the gene regions which inlude seqname, start,end and geneName. That’s enough.
Process gtf file:
library(rtracklayer)
library(tidyverse)
library(ggcirclize)
gtf <- import.gff("Homo_sapiens.GRCh38.110.chr.gtf.gz") %>% as.data.frame()
sample_genes <- sample(unique(gtf$gene_name),40,replace = F)
genes_region <- gtf %>% filter(gene_name %in% sample_genes & type == "gene") %>%
mutate(seqnames = paste0("chr",seqnames))
# check
head(genes_region[,1:7],3)
# seqnames start end width strand source type
# 1 chr1 17066761 17119451 52691 - ensembl_havana gene
# 2 chr1 111490317 111490423 107 - ensembl gene
# 3 chr1 161623196 161631963 8768 - ensembl_havana gene
sample_genes2 <- sample(unique(gtf$gene_name),40,replace = F)
genes_region2 <- gtf %>% filter(gene_name %in% sample_genes2 & type == "gene") %>%
mutate(seqnames = paste0("chr",seqnames))Plot:
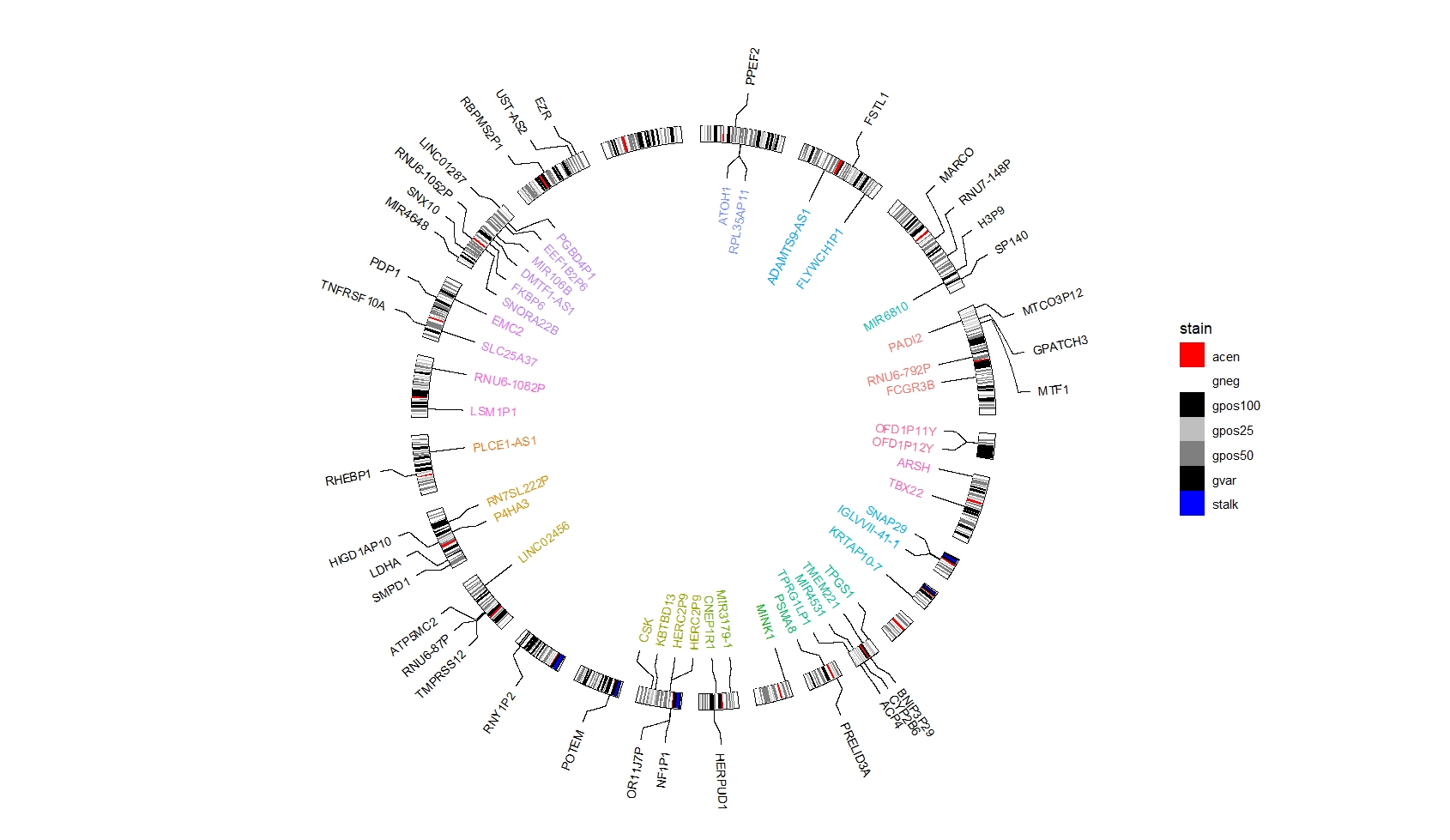
data("hg38_chrom_info")
cytoband_hg38 <- hg38_chrom_info$cytoband
ggcirclize(cytoband_hg38,
aes(end = 360,genome = "hg38",
chr = chr,gstart = start,gend = end)) +
ggcirclize::geom_trackgenomicrect(aes(r0 = 0.8,r1 = 0.85,fill = stain),
color = NA,add.xaxis = F,strip.label = F) +
scale_fill_manual(values = c("gneg" = "white","gpos25" = "grey75","gpos50" = "grey50",
"gpos100" = "black","gvar" = "black","acen" = "red",
"stalk" = "blue")) +
ggcirclize::geom_trackgenomiclabel2(data = genes_region,
aes(r0 = 0.7,r1 = 0.7,label = gene_name,
chr = seqnames,gstart = start,gend = end,
color = seqnames),
keep.all.chrom = T,
strip.label = F) +
ggcirclize::geom_trackgenomiclabel(data = genes_region2,
aes(r0 = 0.95,r1 = 0.95,label = gene_name,
chr = seqnames,gstart = start,gend = end),
link_pos = "bottom",
keep.all.chrom = T,
strip.label = F)