Chapter 8 Nested zooming
To visualize or highlight some specific regions on genome, a convenient way is to draw a new track or sector to show the target region. geom_genomicNestedZoom can zoom target regions with different geom layers.
8.1 Intro
set.seed(111)
bed = generateRandomBed(nr = 600)
zoom <- generateRandomBed(nr = 20)[,-4]
# check
head(zoom,3)
# chr start end
# 1 chr1 52965429 124887955
# 2 chr1 168604490 233330834
# 3 chr2 2089280 5242329
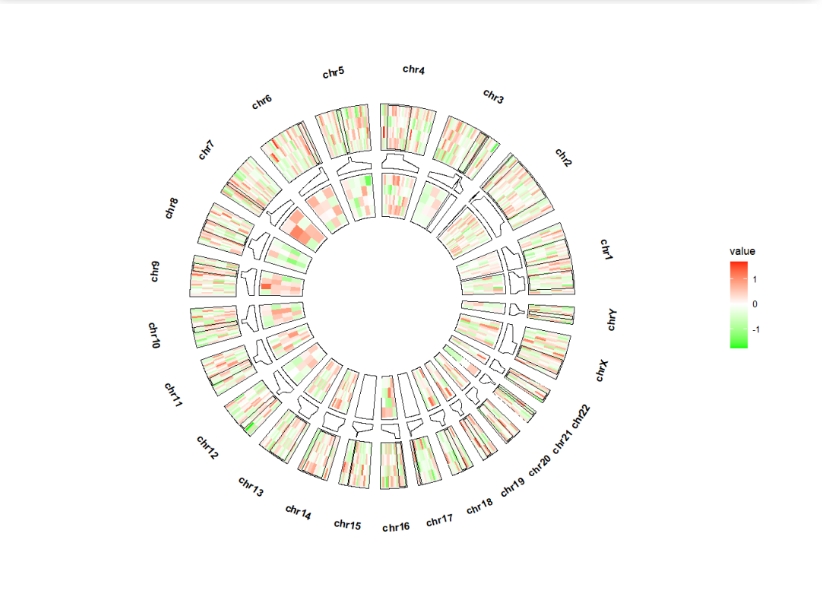
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19")) +
geom_genomicNestedZoom(zoom_data = zoom)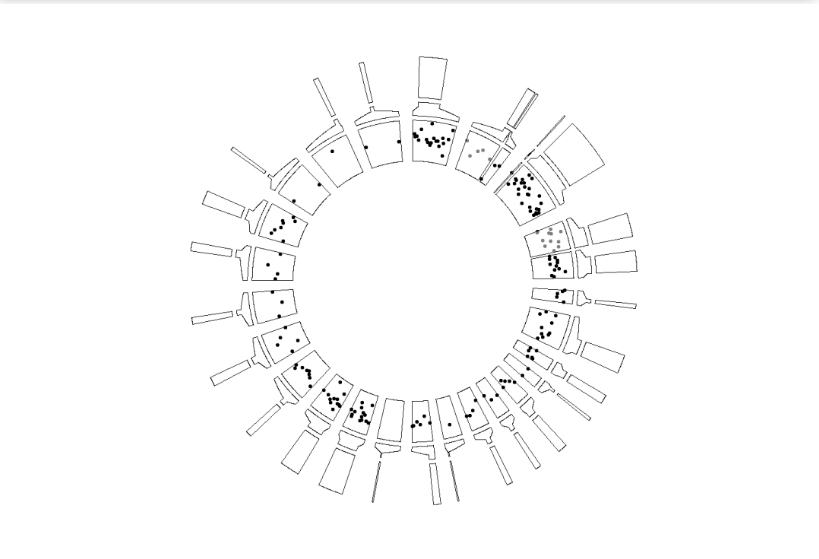
Add origin point layer:
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19",
color = value1)) +
geom_trackgenomicpoint(add.xaxis = F) +
geom_genomicNestedZoom(zoom_data = zoom)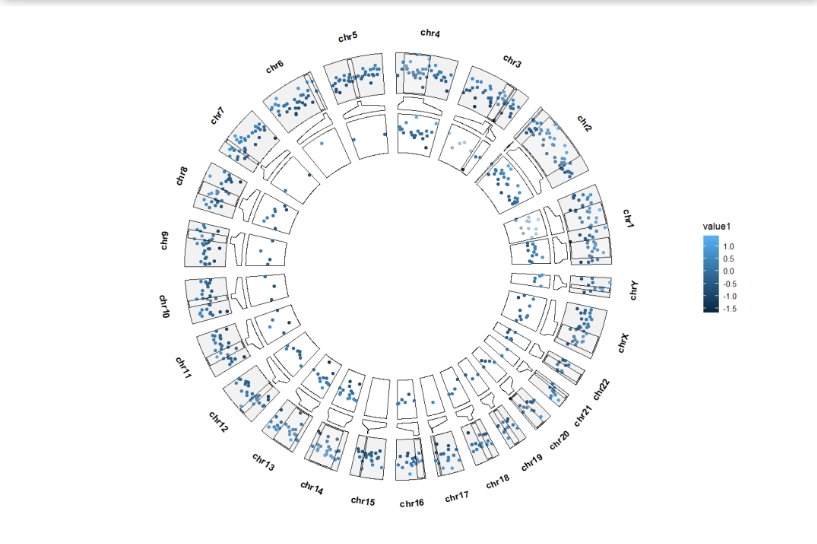
zoom.free.sec can free the zoomed sector size rather than fixed size which is restricted by their origin sector:
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19",
color = value1)) +
geom_trackgenomicpoint(add.xaxis = F) +
geom_genomicNestedZoom(zoom_data = zoom,nested_fill = rand_color(24),
zoom.free.sec = T)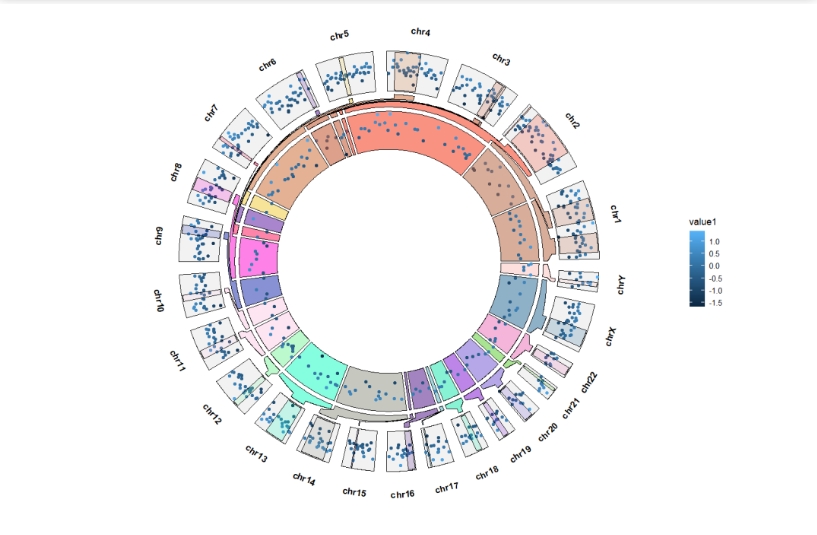
Adjust the zoom sector gap space:
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19",
color = value1)) +
geom_trackgenomicpoint(add.xaxis = F) +
geom_genomicNestedZoom(zoom_data = zoom,nested_fill = rand_color(24),
zoom.free.sec = T,
sub.zoom.gap = 3)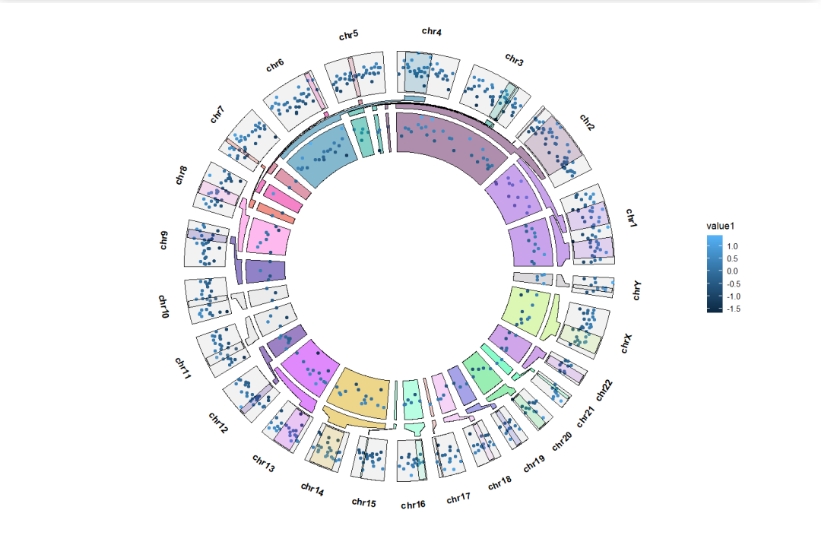
Extend the sector start and end degree:
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19",
color = value1)) +
geom_trackgenomicpoint(add.xaxis = F,sector.gap = 10) +
geom_genomicNestedZoom(zoom_data = zoom,
sector.gap = 10,
extend.start = 5,
extend.end = 5)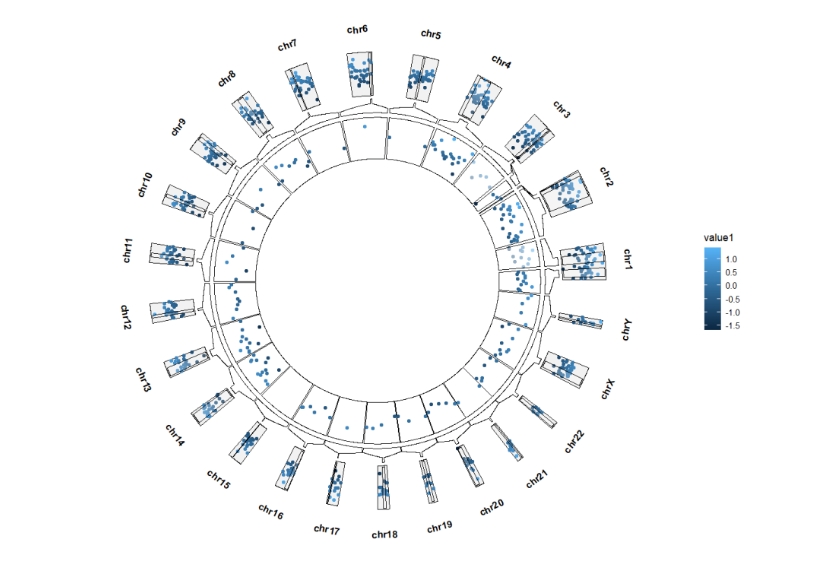
Adjust the zoom track position:
ggcirclize(bed,aes(end = 360,r0 = 0.5,r1 = 0.7,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19",
color = value1)) +
geom_trackgenomicpoint(add.xaxis = F,sector.gap = 5,
strip.label.pos = "bottom",strip.label.space = 0.05) +
geom_genomicNestedZoom(zoom_data = zoom,
sector.gap = 5,
extend.start = 2.5,
extend.end = 2.5,
zoom_pos = "top",
nested_fill = rand_color(24))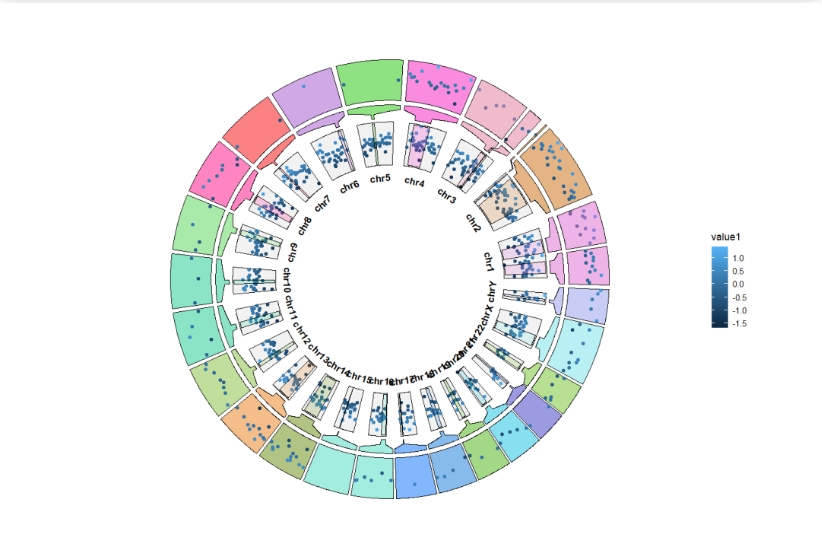
8.2 Zooming with other layers
The default geom is point:
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,
genome = "hg19")) +
geom_trackgenomicline(add.xaxis = F) +
geom_genomicNestedZoom(zoom_data = zoom)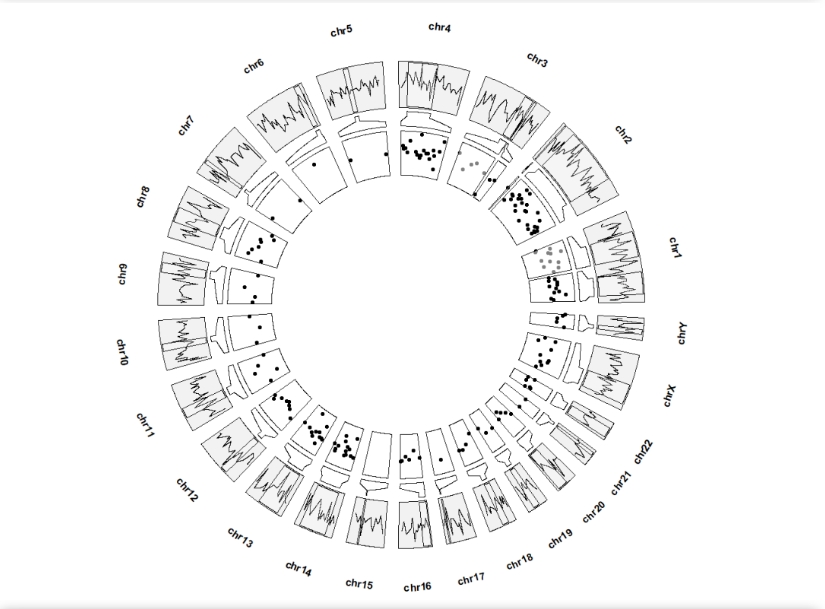
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,color = chr,
genome = "hg19")) +
geom_trackgenomicline(add.xaxis = F,show.legend = F) +
geom_genomicNestedZoom(zoom_data = zoom,
geom = "line",
show.legend = F) +
geom_trackgenomicpoint(aes(r0 = 0.4,r1 = 0.5),
add.xaxis = F,show.legend = F,strip.label = F)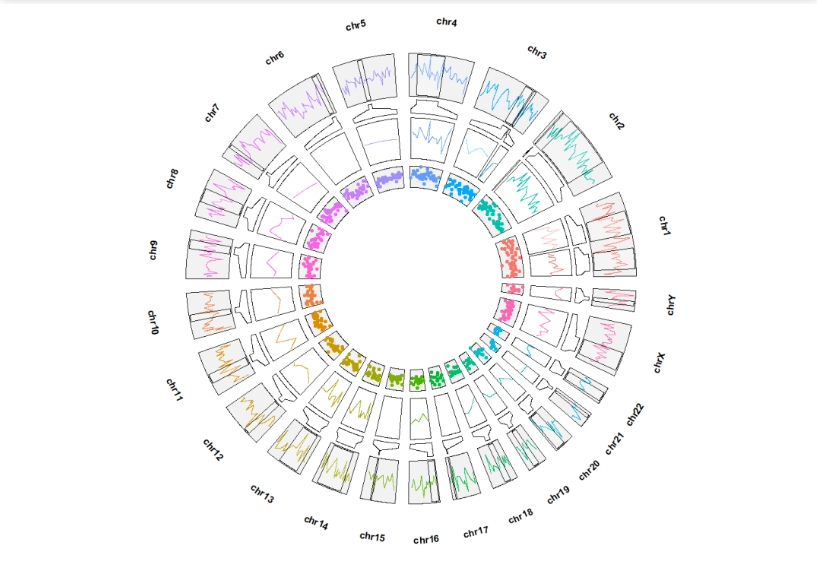
geom_trackgenomicarea:
ggcirclize(bed,aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
value = value1,fill = chr,
genome = "hg19")) +
geom_trackgenomicarea(add.xaxis = F,show.legend = F) +
geom_genomicNestedZoom(zoom_data = zoom,color = "black",show.legend = F,
geom = "area")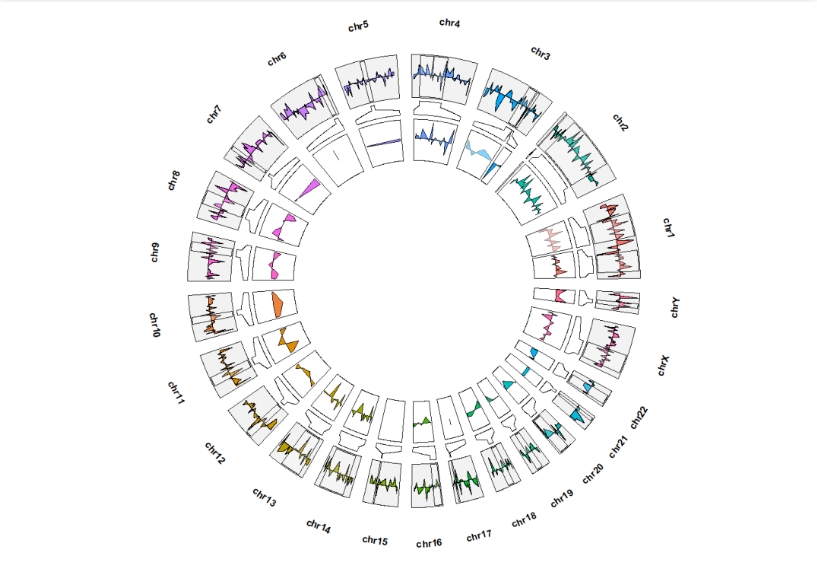
geom_trackgenomicrect:
ggcirclize(cytoband_hg19,
aes(end = 360,r0 = 0.8,r1 = 1,
chr = chr,gstart = start,gend = end,
genome = "hg19")) +
geom_trackgenomicrect(aes(fill = stain,),add.xaxis = F,color = NA) +
scale_fill_manual(values = c("gneg" = "white","gpos25" = "grey75","gpos50" = "grey50",
"gpos100" = "black","gvar" = "black","acen" = "red",
"stalk" = "blue")) +
geom_genomicNestedZoom(zoom_data = zoom,color = "black",
geom = "rect")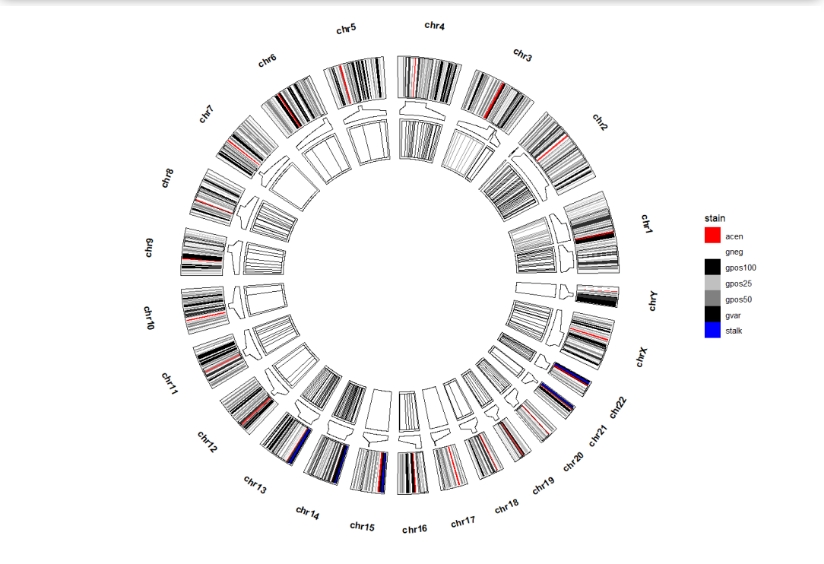
geom_trackgenomictile:
bed = generateRandomBed(nr = 500, nc = 4)
bed_long <- bedMatTolong(bed)
ggcirclize(bed_long,aes(end = 360,r0 = 0.6,r1 = 0.8,
chr = chr,gstart = start,gend = end,
genome = "hg19",
x = x,y = y,fill = value)) +
geom_trackgenomictile(strip.label = T,color = NA) +
scale_fill_gradient2(low = "green",mid = "white",high = "red",midpoint = 0) +
geom_genomicNestedZoom(zoom_data = zoom,geom = "tile",color = NA)