4 Align on transcriptome
This chapter we will describe the total ribo-seq workflow with aligning reads on transcriptome. The test dataset is also same with GSE157519(Adaptive translational pausing is a hallmark of the cellular response to severe environmental stress).
Here we directly use the clean fastq files with removed rRNA and tRNA in the former chapter.
4.1 Extract longest transcript sequence
We need to extract the longest transcript sequence and index for them:
# ==============================================================================
# extract longest transcript fasta
# ==============================================================================
library(BioSeqUtils)
library(dplyr)
# make object
mytest <- loadGenomeGTF(gtfPath = "Mus_musculus.GRCm38.102.gtf.gz",filterProtein = T)
# all gene
gene <- unique(mytest@gtf$gene_name)
# get transcript info
rt <- getTransInfo(object = mytest,geneName = gene,
selecType = "lcds",topN = 1)
# add id
rt <- rt %>%
mutate(seq_id = paste(gene_name,gene_id,transcript_id,
`5UTR` + 1,`5UTR` + CDS,exon,sep = "|"))
# using python code
system.time(pyExtractSeq(gtf_file = "Mus_musculus.GRCm38.102.gtf.gz",
genome_file = "Mus_musculus.GRCm38.dna.primary_assembly.fa",
transcript_id = rt$transcript_id,
new_id = rt$seq_id,
type = "exon",
out_file = "GRCm38_longest_transcript.fasta"))
# check
system("head GRCm38_longest_transcript.fasta")
# >0610009B22Rik|ENSMUSG00000007777|ENSMUST00000109098|209|628|892
# GCAGCCTTGCTCAGAGACGCATGTGCGCATGCCCGGTCGACTGAGCTGCCCTGATCCAAGATGGCGGCCGGACCGGGGCT
# GGGGACGGGCTTAGCGCGTTACTGAGCATGCTCAGTCTTCCGTCACTTGCGTCAGGTCGGCAGCCGGCGCAGGCCGCTCA
# GTCCTTCCCGGAGAGGCGGGAACATGAGCCACATATAGACAGAAGACAATGTCTGGGAGCTTCTACTTCGTAATTGTTGG
# CCACCATGATAATCCGGTTTTTGAAATGGAATTTTTGCCAGCTGGGAAAGCAGAATCTAAAGATGAACACCGTCATCTGA
# ACCAGTTCATAGCTCATGCTGCTCTGGACCTCGTCGACGAAAACATGTGGCTCTCCAACAACATGTACTTAAAAACTGTG
# GACAAATTCAATGAGTGGTTCGTCTCGGCGTTCGTCACGGCTGGGCACATGCGGCTCATCATGCTGCATGACGTGAGGCA
# CGAGGATGGCATCAAGAACTTCTTCACTGACGTCTACGACTTATACATCAAATTTGCCATGAATCCCTTTTATGAACCCA
# ATTCTCCTATTCGATCGAGTGCATTCGAAAGAAAAGTTCAGTTTCTTGGGAAGAAACATCTTTTAAACTAAATGCAGAAA
# AACTTCAGAATCACAGTGGGGTGTGCTCAGCAGTGGATGTATTGTAAATTACTTGATAAAGTAGCCTAGCAAACTCTTTA
# [1] 04.2 Build index for transcriptome
Then we buid index for the longest transcript fasta:
# ==============================================================================
# build index
# ==============================================================================
reference_index_build(reference_file = "GRCm38_longest_transcript.fasta",
prefix = "GRCm38_ref",
threads = 24)
# ┌────────────────────────────────────────────────┐
# │ │
# │ Building index for reference has finished! │
# │ │
# └────────────────────────────────────────────────┘4.3 Remove rRNA and tRNA contamination
Because we have done this before in mapping on genome chapter, so we skip this step.
4.4 Map to transcriptome
# ==============================================================================
# map to genome
# ==============================================================================
dir.create("4.map-data")
# batch map to genome
fq_file1 <- list.files("../1.mapping_to_genome/3.rmtrRNA-data/",pattern = ".fq",full.names = T)
output_file <- output_file <- c("ribo_nromal_rep1","ribo_700_2h_rep1","ribo_700_2h_r2h_rep1",
"ribo_nromal_rep2","ribo_700_2h_rep2","ribo_700_2h_r2h_rep2",
"rna_nromal_rep1","rna_700_2h_rep1","rna_700_2h_r2h_rep1",
"rna_nromal_rep2","rna_700_2h_rep2","rna_700_2h_r2h_rep2")
batch_hisat_align(index = "0.index-data/ref-index/GRCm38_ref",
fq_file1 = fq_file1,
output_dir = "4.map-data/",
output_file = output_file,
threads = 24,hisat2_params = list(k = 1))
# ▶ ribo_nromal_rep1 has been processed!
# ▶ ribo_700_2h_rep1 has been processed!
# ▶ ribo_700_2h_r2h_rep1 has been processed!
# ▶ ribo_nromal_rep2 has been processed!
# ▶ ribo_700_2h_rep2 has been processed!
# ▶ ribo_700_2h_r2h_rep2 has been processed!
# ...Plot mapinfo barplot:
# plot mapping info
map_info_geome <- list.files("4.map-data/",pattern = "mapinfo.txt",full.names = TRUE)
plot_mapinfo(mapinfo_file = map_info_geome,
file_name = sapply(strsplit(map_info_geome,split = "_mapinfo.txt|/"), "[",2))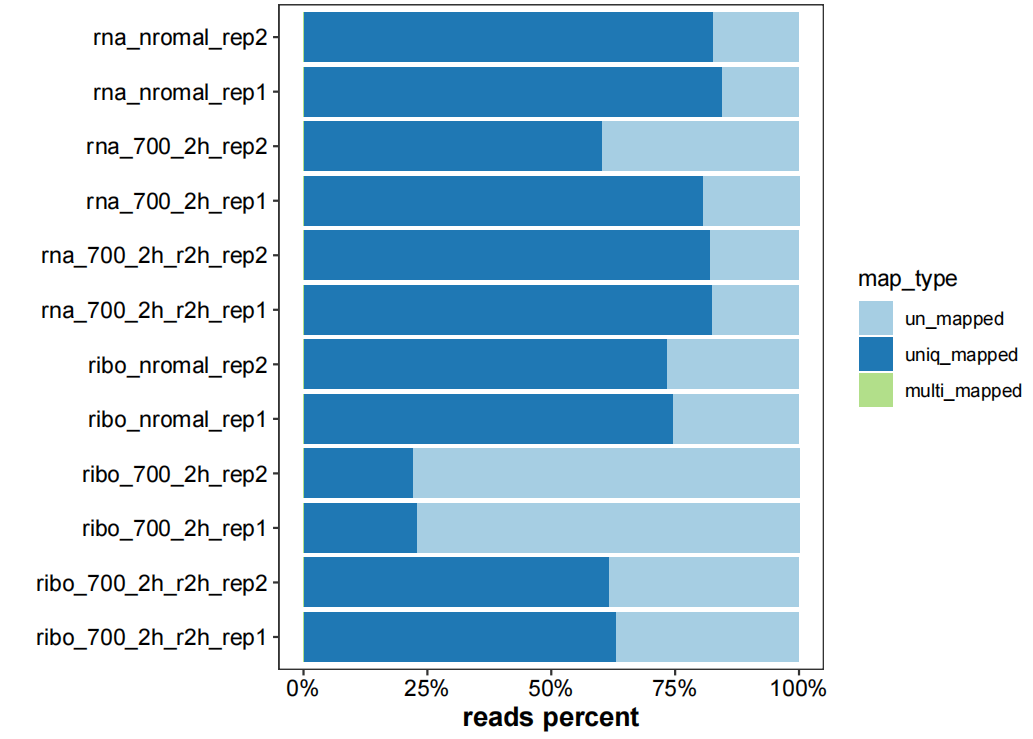
Plot with a table:
plot_mapinfo(mapinfo_file = map_info_geome,
file_name = sapply(strsplit(map_info_geome,split = "_mapinfo.txt|/"), "[",2),
plot_type = "table")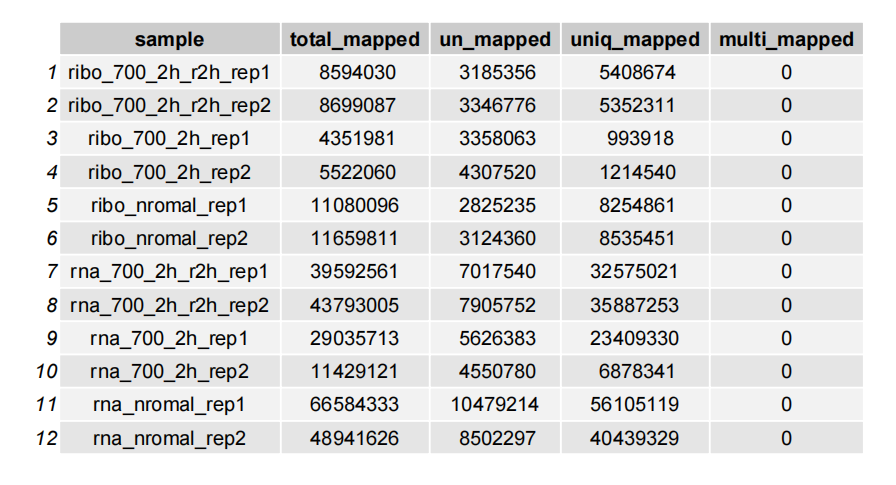
4.5 Sam to bam
Covert the sam files into bam format:
# ==============================================================================
# sam to bam
# ==============================================================================
dir.create("4.map-data/bam-data",recursive = TRUE)
# convert sam to bam
batch_sam2bam(sam_file = paste("4.map-data/",output_file,".sam",sep = ""),
bam_file = paste("4.map-data/bam-data/",output_file,sep = ""))
# ▶ 4.map-data/rna_nromal_rep1.sam has been processed!
# [bam_sort_core] merging from 12 files and 1 in-memory blocks...
# ▶ 4.map-data/rna_700_2h_rep1.sam has been processed!
# [bam_sort_core] merging from 17 files and 1 in-memory blocks...
# ▶ 4.map-data/rna_700_2h_r2h_rep1.sam has been processed!
# [bam_sort_core] merging from 21 files and 1 in-memory blocks...
# ...4.6 Ribo-seq QC check
4.6.1 prepare longest transcript
First we should prepare the longest transcript for each protein gene with gene annotation file:
dir.create("5.analysis-data")
setwd("5.analysis-data")
# ==============================================================================
# 1_prepare gene annotation file
# ==============================================================================
pre_longest_trans_info(gtf_file = "../Mus_musculus.GRCm38.102.gtf.gz",
out_file = "longest_info.txt")
# ================================ job finished.4.6.2 prepare QC data
pre_qc_data will produce QC data for ribo-seq sam files:
# ==============================================================================
# 2_prepare Ribo QC data
# ==============================================================================
sam_file = list.files("../4.map-data/",pattern = "^ribo.*sam$",full.names = TRUE)
sam_file
# [1] "../4.map-data/ribo_700_2h_r2h_rep1.sam" "../4.map-data/ribo_700_2h_r2h_rep2.sam"
# [3] "../4.map-data/ribo_700_2h_rep1.sam" "../4.map-data/ribo_700_2h_rep2.sam"
# [5] "../4.map-data/ribo_nromal_rep1.sam" "../4.map-data/ribo_nromal_rep2.sam"
sample_name <- sapply(strsplit(list.files("../4.map-data/",pattern = "^ribo.*sam$"),
split = ".sam"), "[",1)
sample_name
# [1] "ribo_700_2h_r2h_rep1" "ribo_700_2h_r2h_rep2" "ribo_700_2h_rep1"
# [4] "ribo_700_2h_rep2" "ribo_nromal_rep1" "ribo_nromal_rep2"
# run
pre_qc_data(sam_file = sam_file,
out_file = paste(sample_name,".qc.txt",sep = ""),
mapping_type = "transcriptome",
seq_type = "singleEnd")
# ../4.map-data/ribo_700_2h_r2h_rep1.sam has been processed!
# ../4.map-data/ribo_700_2h_r2h_rep2.sam has been processed!
# ../4.map-data/ribo_700_2h_rep1.sam has been processed!
# ...4.6.3 load qc data
load_qc_data will read all the qc data into R and return a data frame:
# load qc data
qc_df <- load_qc_data()
qc_df <- qc_df |> dplyr::filter(length >= 20 & length <= 35)
# QC input files:
# ribo_700_2h_r2h_rep1.qc.txt
# ribo_700_2h_r2h_rep2.qc.txt
# ribo_700_2h_rep1.qc.txt
# ribo_700_2h_rep2.qc.txt
# ribo_nromal_rep1.qc.txt
# ribo_nromal_rep2.qc.txt
# check
head(qc_df,3)
# length framest relst framesp relsp feature counts sample group
# 1 27 0 825 0 -168 3 1 ribo_700_2h_r2h_rep1 NA
# 2 30 2 1916 1 -694 3 1 ribo_700_2h_r2h_rep1 NA
# 3 30 0 1401 0 -21 3 7 ribo_700_2h_r2h_rep1 NA
qc_df$length <- factor(qc_df$length)
qc_df$sample <- factor(qc_df$sample,
levels = c("ribo_nromal_rep1","ribo_700_2h_rep1","ribo_700_2h_r2h_rep1",
"ribo_nromal_rep2","ribo_700_2h_rep2","ribo_700_2h_r2h_rep2"))4.6.4 qc visualization
Now plot the qc data with multiple kinds of graph, first check the length distribution of fragments:
# length distribution
qc_plot(qc_data = qc_df,type = "length",facet_wrap_list = list(ncol = 3))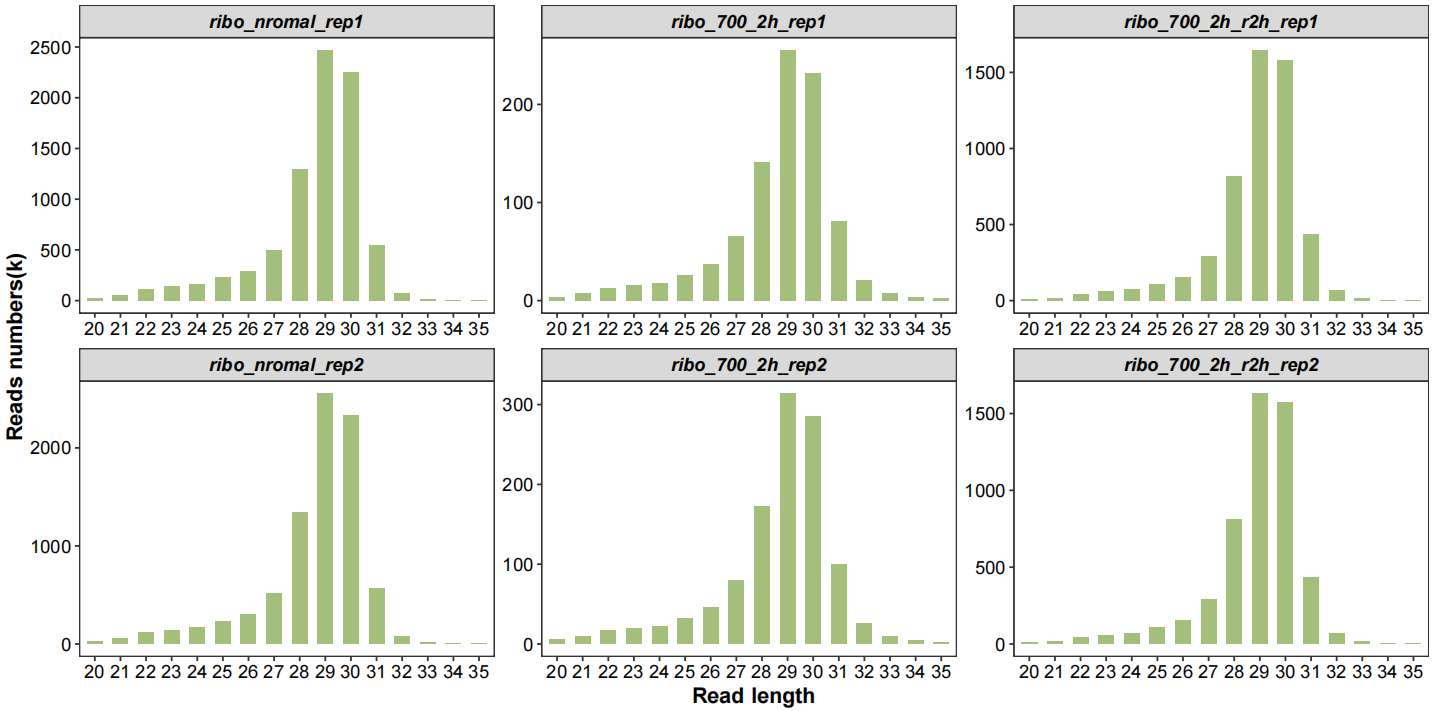
Frames with length distribution:
# length with frame
qc_plot(qc_data = qc_df,type = "length_frame",facet_wrap_list = list(ncol = 3))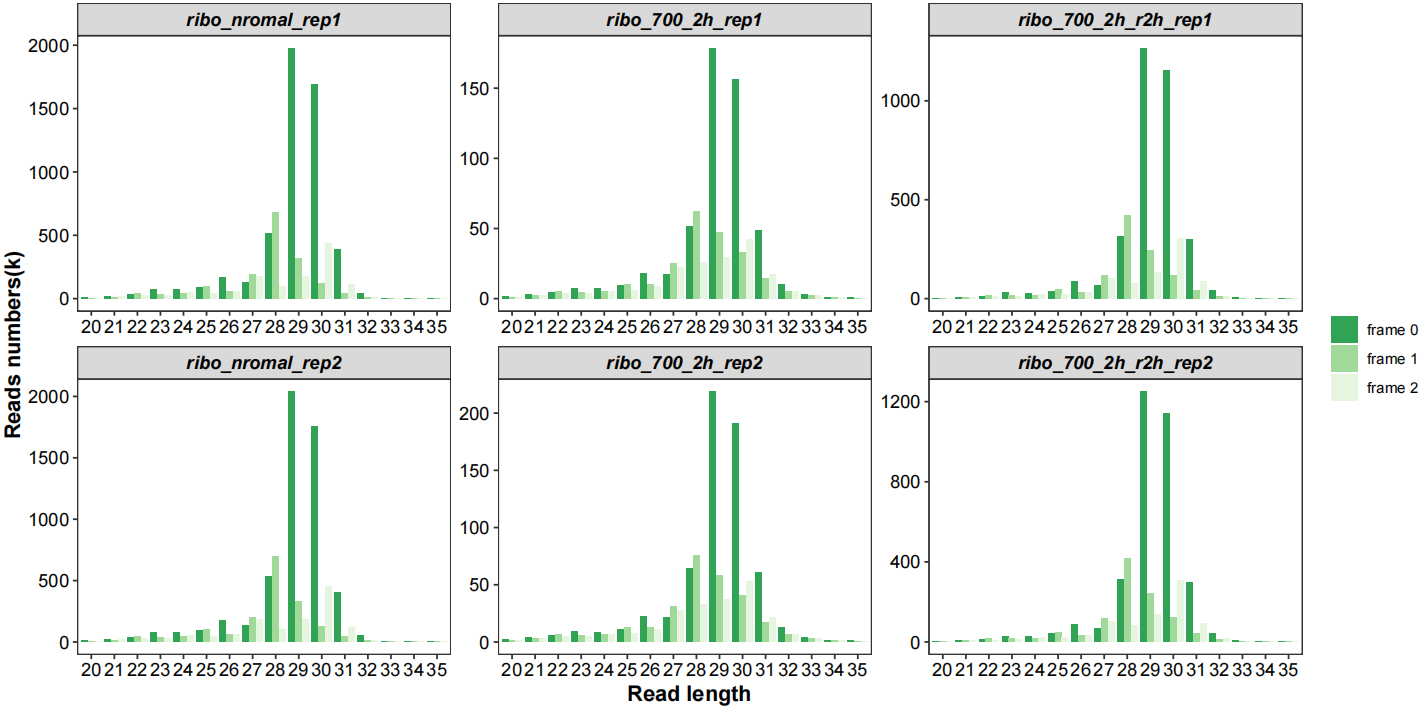
Different region features:
# region features
qc_plot(qc_data = qc_df,type = "feature",facet_wrap_list = list(ncol = 3))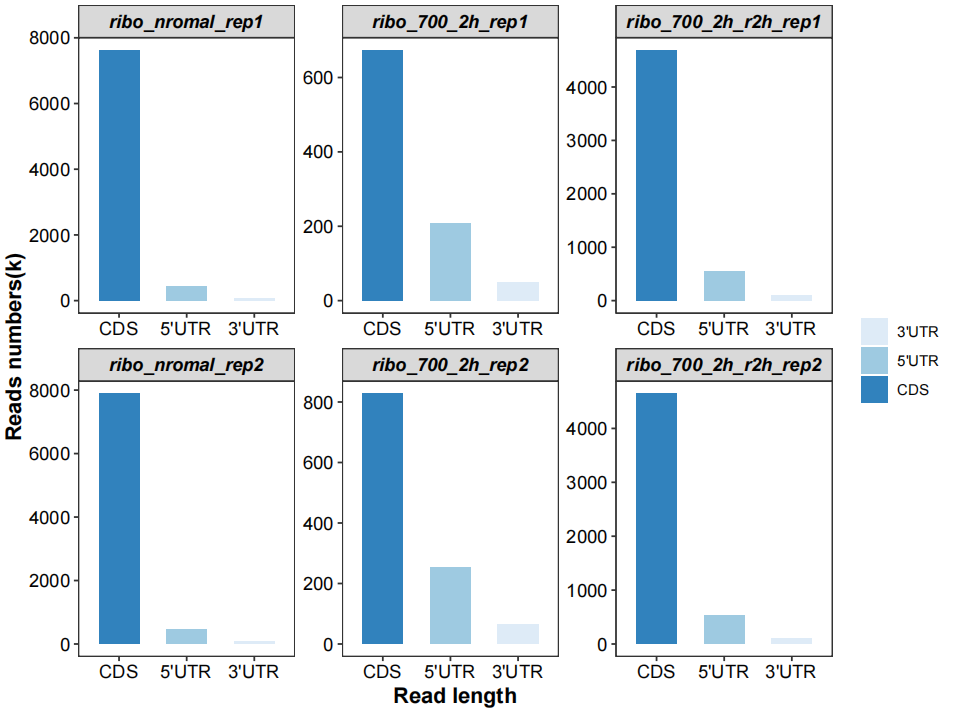
All frames ratio:
# all frames
qc_plot(qc_data = qc_df,type = "frame",facet_wrap_list = list(ncol = 3))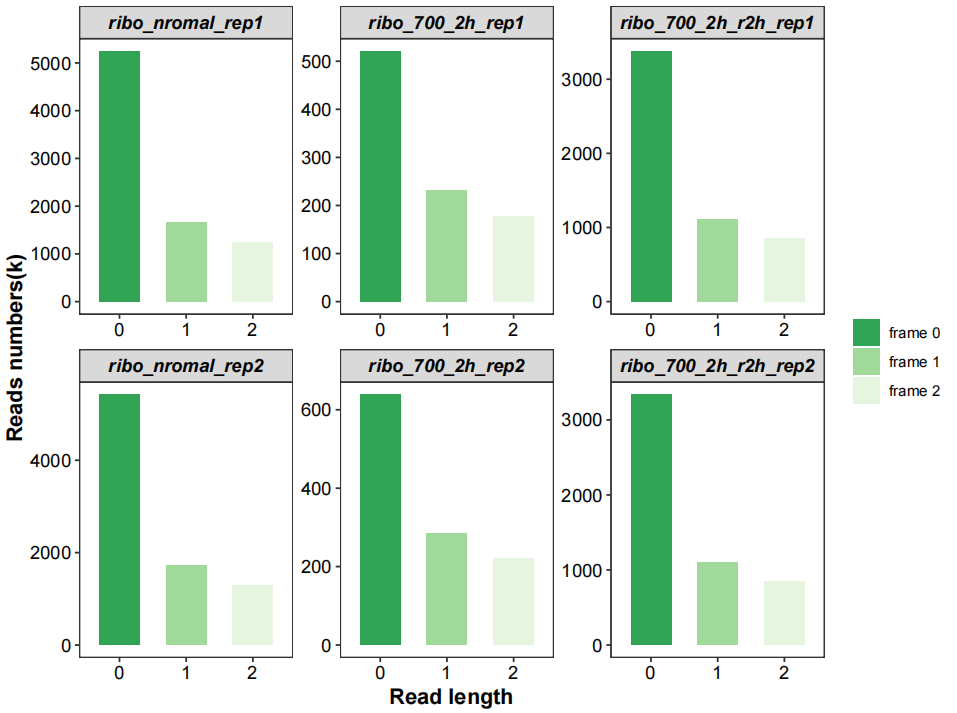
Realtive to start codon with periodicity:
# relative to start/stop site 5x10
rel_to_start_stop(qc_data = qc_df,type = "relst",
facet_wrap_list = list(ncol = 3,scales = "fixed"))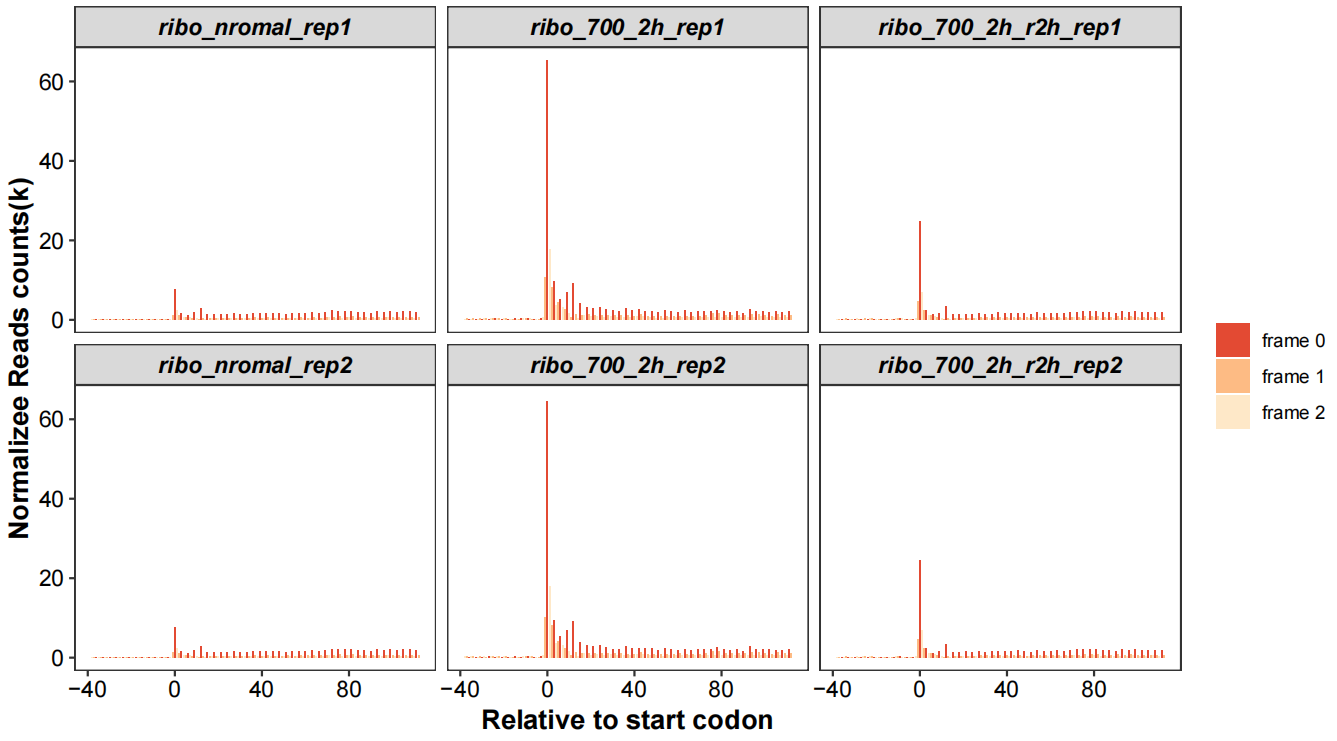
Realtive to stop codon with periodicity:
rel_to_start_stop(qc_data = qc_df,type = "relsp",
facet_wrap_list = list(ncol = 3,scales = "fixed"))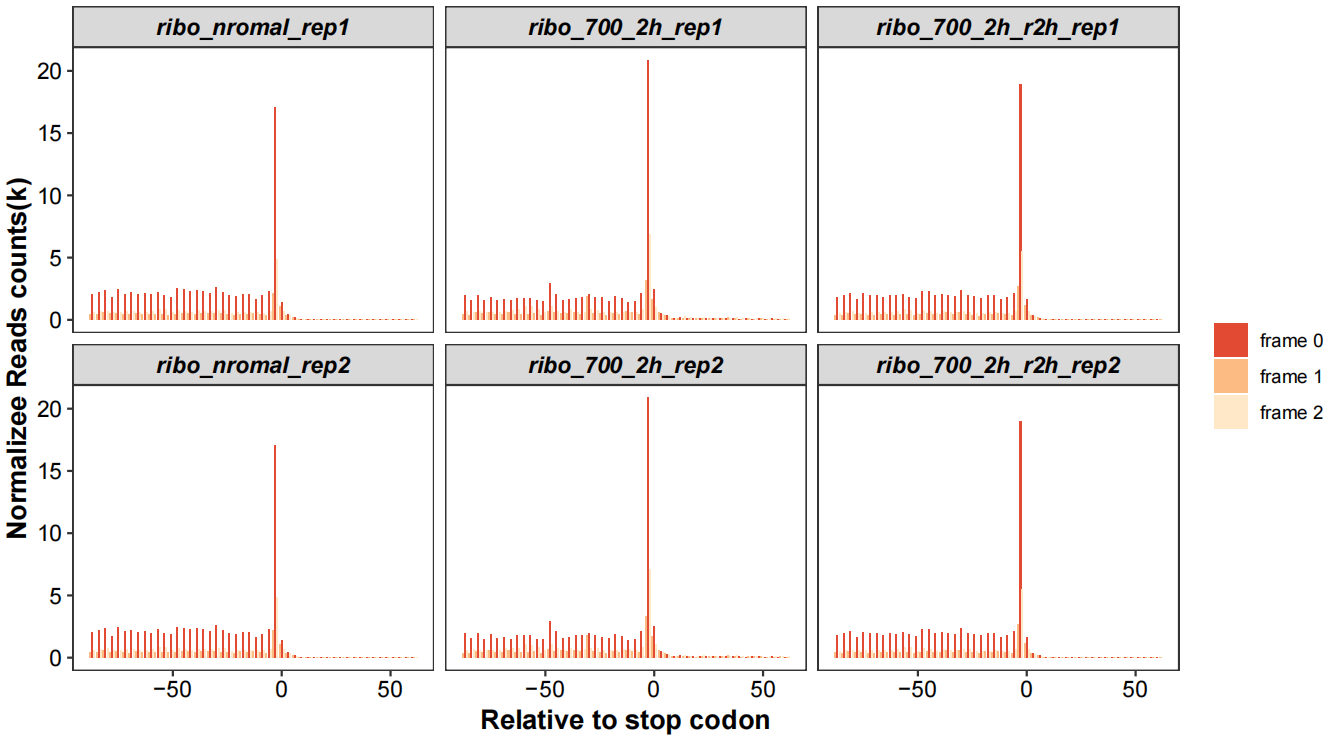
4.8 Calculate Ribo density and RNA coverage
# ==============================================================================
# 3_prepare Ribo density and RNA coverage data
# ==============================================================================
# calculate Ribo density
pre_ribo_density_data(sam_file = sam_file,
out_file = paste(sample_name,".density.txt",sep = ""),
mapping_type = "transcriptome")
# ../4.map-data/ribo_700_2h_r2h_rep1.sam has been processed!
# ../4.map-data/ribo_700_2h_r2h_rep2.sam has been processed!
# ../4.map-data/ribo_700_2h_rep1.sam has been processed!
# ...
# calculate RNA coverage
bam_file_rna = list.files("../4.map-data/bam-data",pattern = "^rna.*bam$",full.names = TRUE)
bam_file_rna
# [1] "../4.map-data/bam-data/rna_700_2h_r2h_rep1.bam" "../4.map-data/bam-data/rna_700_2h_r2h_rep2.bam"
# [3] "../4.map-data/bam-data/rna_700_2h_rep1.bam" "../4.map-data/bam-data/rna_700_2h_rep2.bam"
# [5] "../4.map-data/bam-data/rna_nromal_rep1.bam" "../4.map-data/bam-data/rna_nromal_rep2.bam"
sample_name_rna <- sapply(strsplit(list.files("../4.map-data/bam-data",pattern = "^rna.*bam$"),
split = ".bam"), "[",1)
sample_name_rna
# [1] "rna_700_2h_r2h_rep1" "rna_700_2h_r2h_rep2" "rna_700_2h_rep1" "rna_700_2h_rep2"
# [5] "rna_nromal_rep1" "rna_nromal_rep2"
pre_rna_coverage_data(bam_file = bam_file_rna,
out_file = paste(sample_name_rna,".coverage.txt",sep = "")
)
# ../4.map-data/bam-data/rna_700_2h_r2h_rep1.bam has been processed!
# ../4.map-data/bam-data/rna_700_2h_r2h_rep2.bam has been processed!
# ../4.map-data/bam-data/rna_700_2h_rep1.bam has been processed!
# ...4.9 Track plot
4.9.1 load track data
# ==============================================================================
# 5_load denisty and coverage data for specific gene
# ==============================================================================
track_df <- load_track_data(ribo_file = paste(sample_name,".density.txt",sep = ""),
rna_file = paste(sample_name_rna,".coverage.txt",sep = ""),
sample_name = paste(rep(c("700_2h_r2h","700_2h","normal"),each = 2),
c("_rep1","_rep2"),sep = ""),
gene_list = c("Ndufb3","Ndufb6","Rps7","Uqcrfs1","Ndufv1","Sat1"),
mapping_type = "transcriptome")
# check
head(track_df,3)
# gene_name trans_id transpos density type sample
# 1 Ndufb3 ENSMUST00000027193 100 0.0528252 ribo 700_2h_r2h_rep1
# 2 Ndufb3 ENSMUST00000027193 102 0.0528252 ribo 700_2h_r2h_rep1
# 3 Ndufb3 ENSMUST00000027193 103 0.0528252 ribo 700_2h_r2h_rep14.9.2 plot tracks
# 6x12
track_plot(signal_data = track_df |> dplyr::filter(gene_name %in% c("Ndufb3","Ndufb6","Rps7")),
gene_anno = "longest_info.txt",
sample_order = c("normal_rep1","normal_rep2","700_2h_rep1","700_2h_rep2",
"700_2h_r2h_rep1","700_2h_r2h_rep2"),
remove_trans_panel_border = T)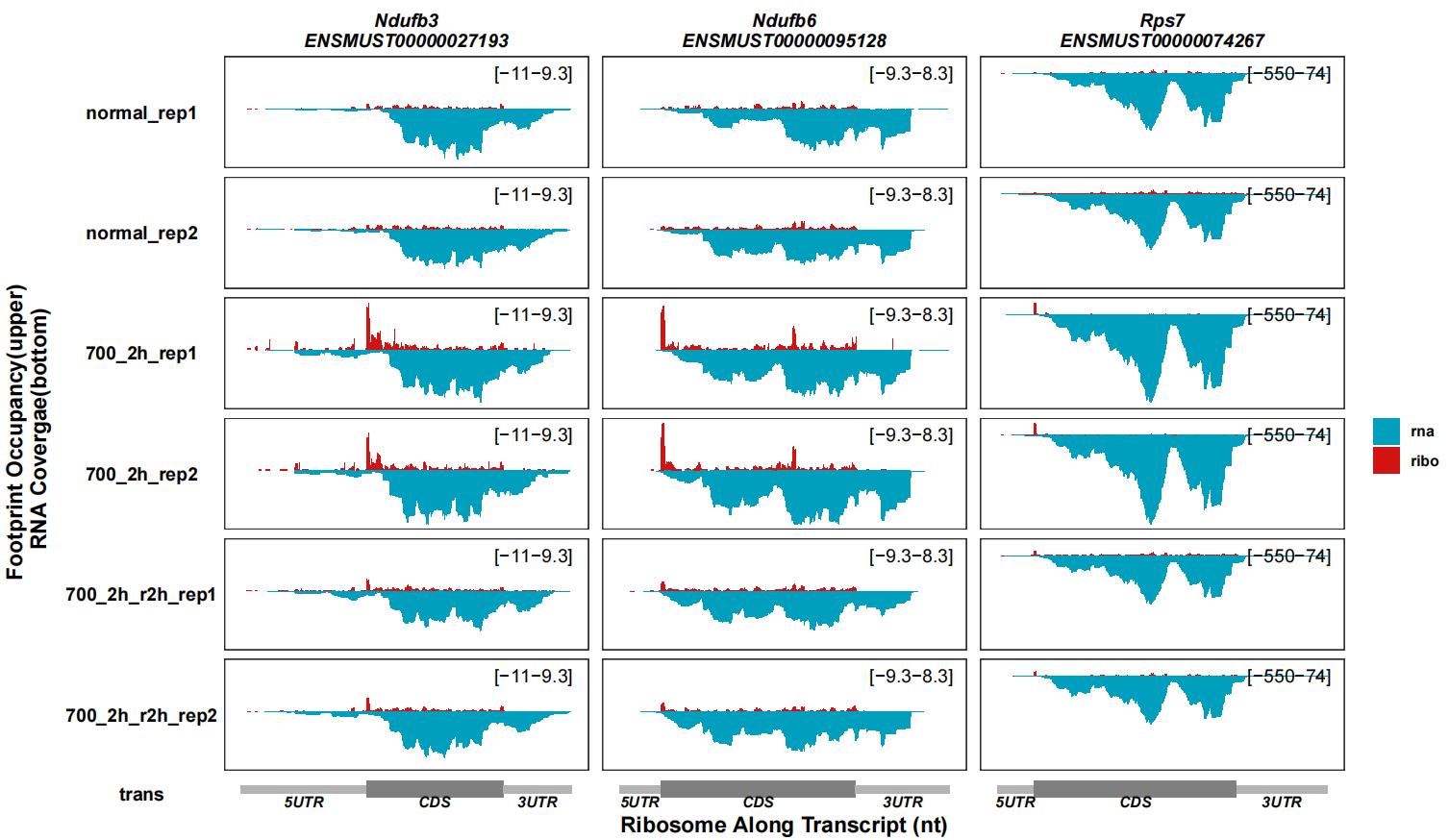
Reverse the RNA coverage track:
# reverse RNA track
track_plot(signal_data = track_df |> dplyr::filter(gene_name %in% c("Ndufb3","Ndufb6","Rps7")),
gene_anno = "longest_info.txt",
sample_order = c("normal_rep1","normal_rep2","700_2h_rep1","700_2h_rep2",
"700_2h_r2h_rep1","700_2h_r2h_rep2"),
remove_trans_panel_border = T,
reverse_rna = F)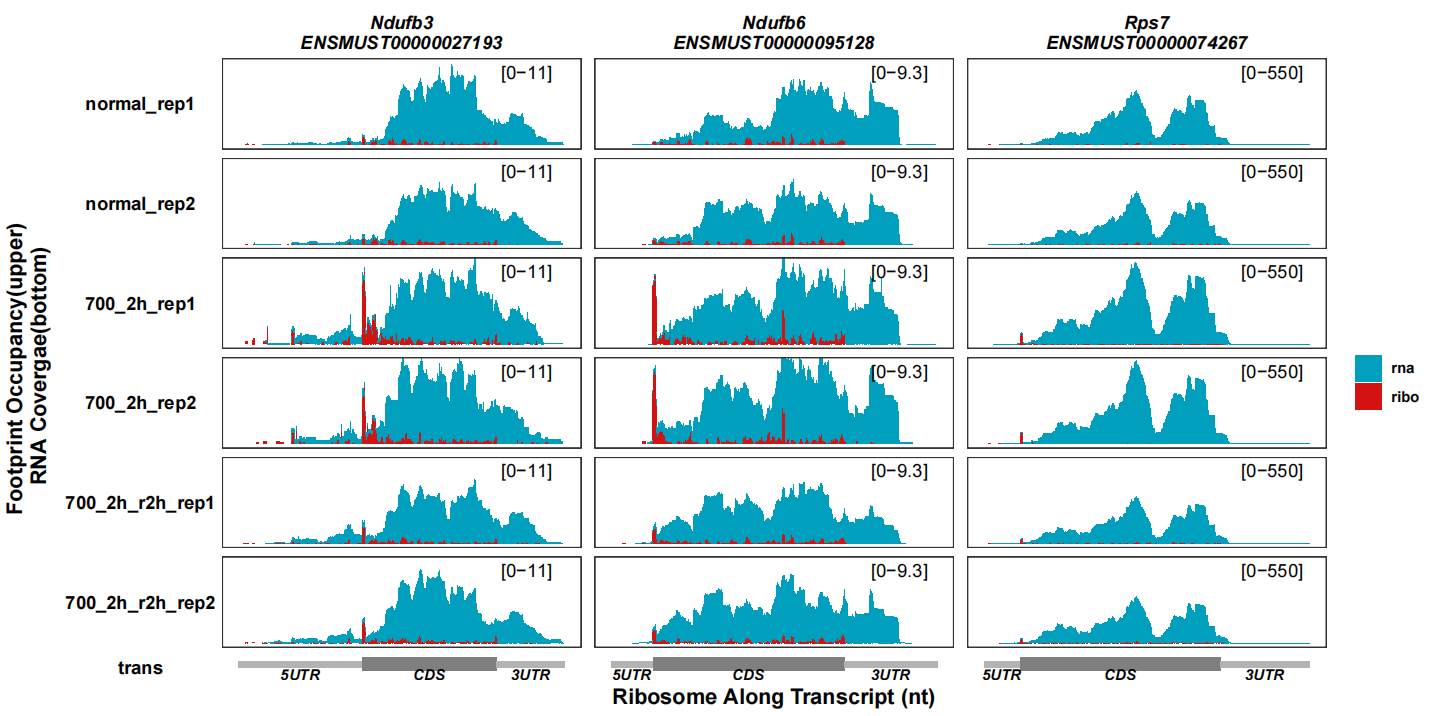
Another three genes:
track_plot(signal_data = track_df |> dplyr::filter(gene_name %in% c("Uqcrfs1","Ndufv1","Sat1")),
gene_anno = "longest_info.txt",
sample_order = c("normal_rep1","normal_rep2","700_2h_rep1","700_2h_rep2",
"700_2h_r2h_rep1","700_2h_r2h_rep2"),
remove_trans_panel_border = T)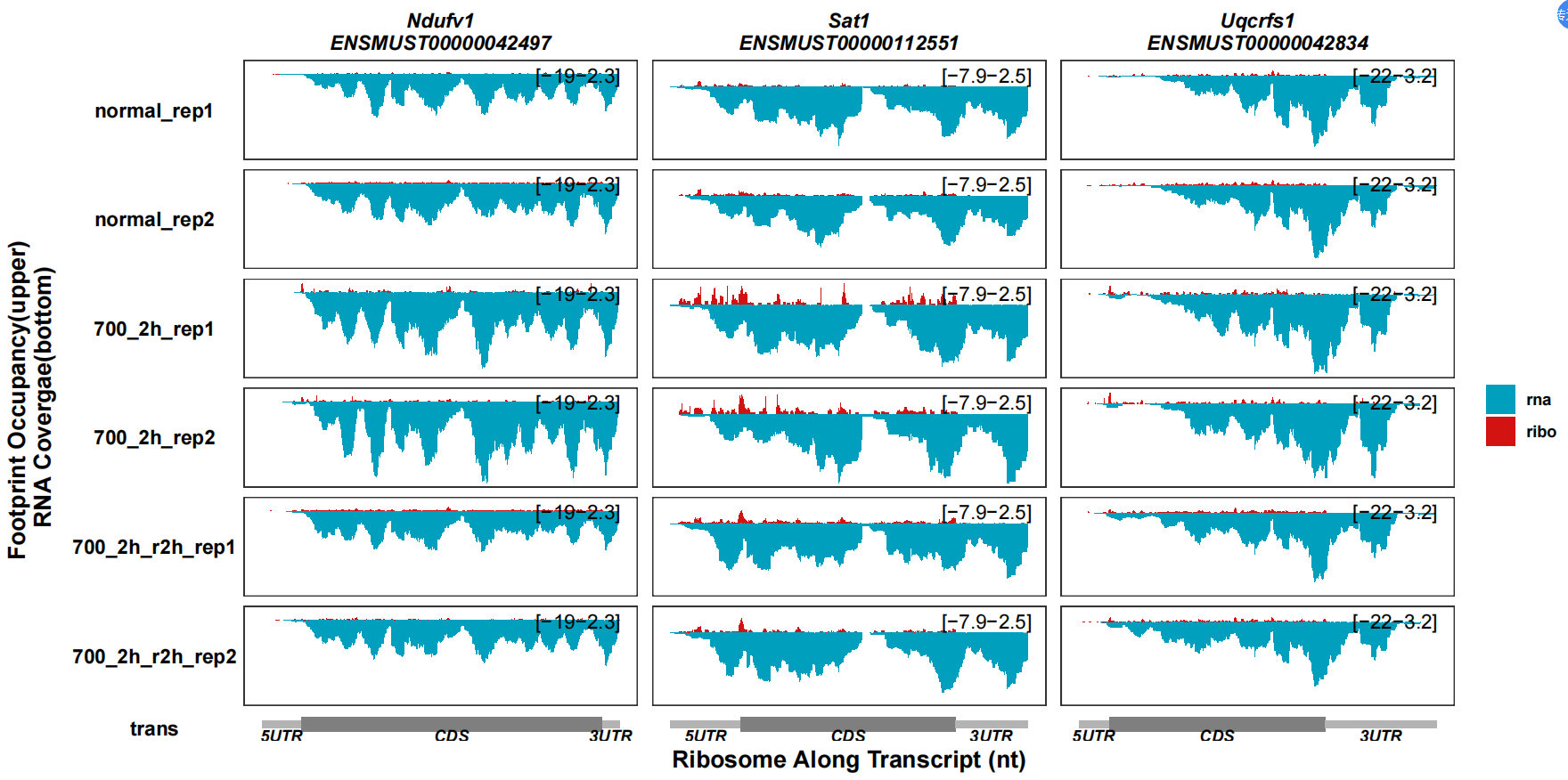
show only ribo tracks:
track_plot(signal_data = track_df |> dplyr::filter(gene_name %in% c("Uqcrfs1","Ndufv1","Sat1")),
gene_anno = "longest_info.txt",
sample_order = c("normal_rep1","normal_rep2","700_2h_rep1","700_2h_rep2",
"700_2h_r2h_rep1","700_2h_r2h_rep2"),
remove_trans_panel_border = T,
show_ribo_only = T)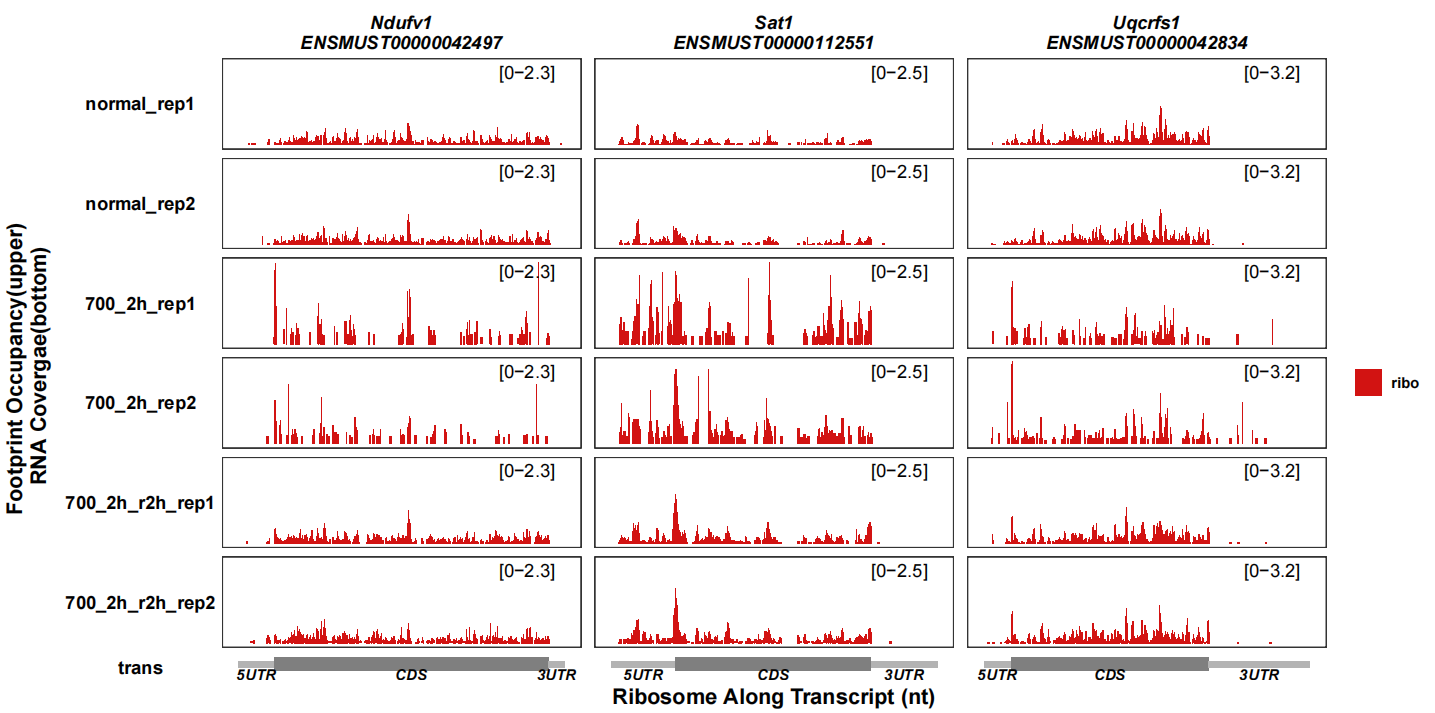
4.10 Metagene plot
Metagene plot shows the average ribosome density relative to start/stop codon:
# ==============================================================================
# 7_metagene plot
# ==============================================================================
# prepare data
pre_metagene_data(density_file = paste(sample_name[c(1,3,5)],".density.txt",sep = ""),
out_file = paste(sample_name[c(1,3,5)],".meta.txt",sep = ""),
mapping_type = "transcriptome")
# 2.density-data/ribo_700_2h_r2h_rep1.density.txt has been processed!
# 2.density-data/ribo_700_2h_rep1.density.txt has been processed!
# 2.density-data/ribo_nromal_rep1.density.txt has been processed!
pre_metagene_data(density_file = paste(sample_name[c(1,3,5)],".density.txt",sep = ""),
out_file = paste(sample_name[c(1,3,5)],".sp.meta.txt",sep = ""),
mapping_type = "transcriptome",
mode = "sp")
# rel2sp
pre_metagene_data(density_file = paste(sample_name[c(1,3,5)],".density.txt",sep = ""),
out_file = paste(sample_name[c(1,3,5)],".sp.meta.txt",sep = ""),
mapping_type = "transcriptome",
mode = "sp")
# load data
meta_df <- load_metagene_data()
# MetaGene input files:
# ribo_700_2h_r2h_rep1.meta.txt
# ribo_700_2h_r2h_rep1.sp.meta.txt
# ribo_700_2h_rep1.meta.txt
# ribo_700_2h_rep1.sp.meta.txt
# ribo_nromal_rep1.meta.txt
# ribo_nromal_rep1.sp.meta.txt
meta_df$group <- rep(rep(c("st","sp"),each = 518),3)Plot:
# plot
library(ggplot2)
meta_df$sample <- factor(meta_df$sample,levels = c("ribo_nromal_rep1","ribo_700_2h_rep1",
"ribo_700_2h_r2h_rep1"))
ggplot(meta_df) +
geom_line(aes(x = pos,y = density,color = sample),linewidth = 1) +
scale_color_brewer(palette = "Set1") +
theme_bw() +
jj_theme() +
ggh4x::facet_grid2(group~sample,scales = "free",independent = "x")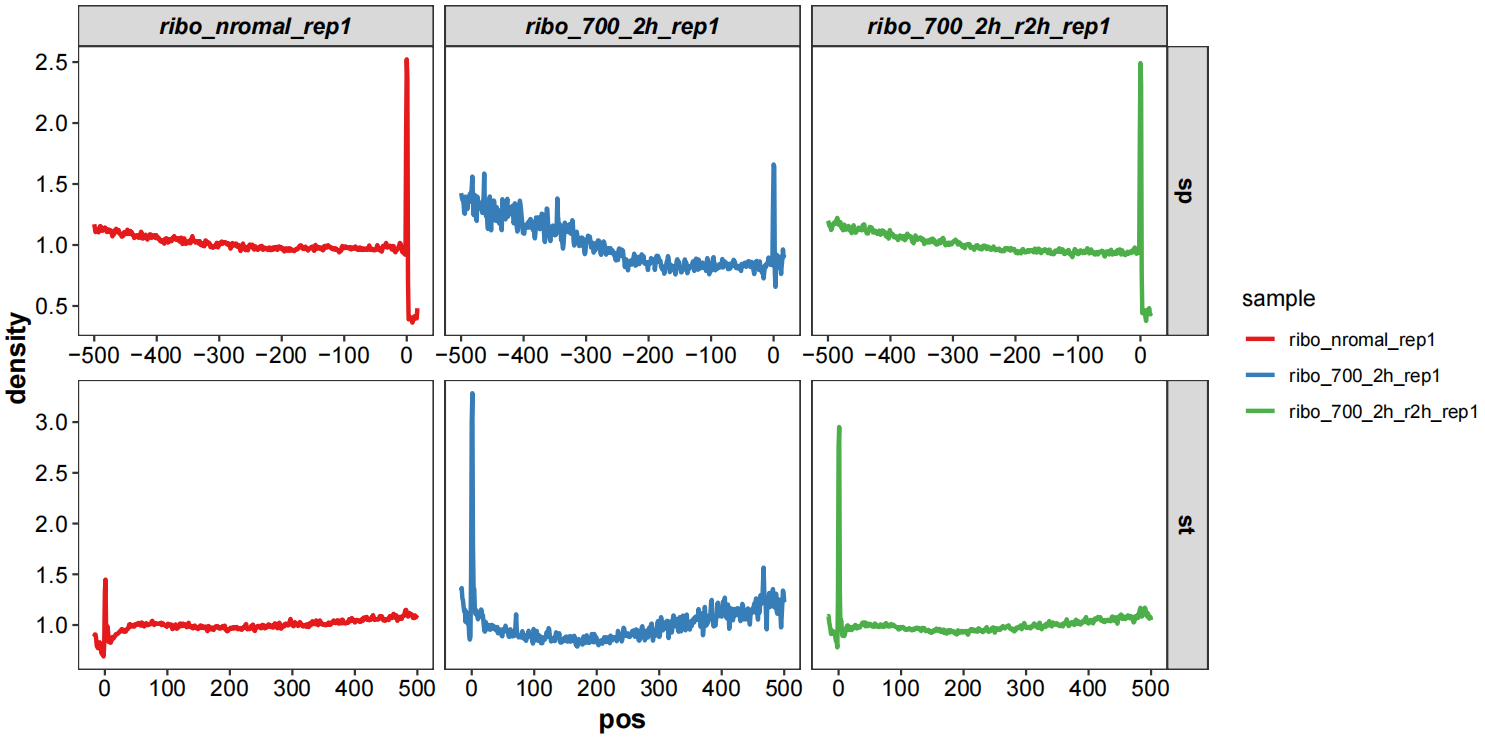
4.11 Quantification
pre_count_tpm_data will calculate genes count and tpm from sam files:
# for Ribo
sam_file = list.files("../4.map-data/",pattern = "^ribo.*sam$",full.names = TRUE)
sample_name <- sapply(strsplit(list.files("../4.map-data/",pattern = "^ribo.*sam$"),
split = ".sam"), "[",1)
# get expression matrix
pre_count_tpm_data(sam_file = sam_file,
out_file = paste(sample_name,".exp.txt",sep = ""),
type = "ribo")
# ../4.map-data/ribo_700_2h_r2h_rep1.sam has been processed!
# ../4.map-data/ribo_700_2h_r2h_rep2.sam has been processed!
# ../4.map-data/ribo_700_2h_rep1.sam has been processed!
# ...
# for RNA
sam_file = list.files("../4.map-data/",pattern = "^rna.*sam$",full.names = TRUE)
sample_name <- sapply(strsplit(list.files("../4.map-data/",pattern = "^rna.*sam$"),
split = ".sam"), "[",1)
# get expression matrix
pre_count_tpm_data(sam_file = sam_file,
out_file = paste(sample_name,".exp.txt",sep = ""),
type = "rna")
# ../4.map-data/rna_700_2h_r2h_rep1.sam has been processed!
# ../4.map-data/rna_700_2h_r2h_rep2.sam has been processed!
# ../4.map-data/rna_700_2h_rep1.sam has been processed!
# ...load_expression_data extract count and tpm matrix from files:
# prepare matrix
exp <- load_expression_data()
# Expression input files:
# ribo_700_2h_r2h_rep1.exp.txt
# ribo_700_2h_r2h_rep2.exp.txt
# ribo_700_2h_rep1.exp.txt
# ribo_700_2h_rep2.exp.txt
# ribo_nromal_rep1.exp.txt
# ribo_nromal_rep2.exp.txt
# rna_700_2h_r2h_rep1.exp.txt
# rna_700_2h_r2h_rep2.exp.txt
# rna_700_2h_rep1.exp.txt
# rna_700_2h_rep2.exp.txt
# rna_nromal_rep1.exp.txt
# rna_nromal_rep2.exp.txt
head(exp$count_matrix[1:3,1:4])
# gene_name ribo_700_2h_r2h_rep1 ribo_700_2h_r2h_rep2 ribo_700_2h_rep1
# 1: 0610009B22Rik 126 124 39
# 2: 0610010F05Rik 81 70 5
# 3: 0610010K14Rik 65 64 2
head(exp$tpm_matrix[1:3,1:4])
# gene_name ribo_700_2h_r2h_rep1 ribo_700_2h_r2h_rep2 ribo_700_2h_rep1
# 1: 0610009B22Rik 51.687154 51.39426 70.832450
# 2: 0610010F05Rik 6.478891 5.65711 1.770685
# 3: 0610010K14Rik 19.544299 19.44319 2.662517