Chapter 5 Heatmap annotation for terms
A heatmap annotation for a specific term which contains a set of core genes can be added under the GSEA plot to illustrate the gene expresion changes between different conditions. We need a normalized expression matrix:
library(clusterProfiler)
library(org.Mm.eg.db)
library(GseaVis)
# load diff
diff <- read.csv('diffdata.csv') %>%
arrange(desc(log2FoldChange))
genelist <- diff$log2FoldChange
names(genelist) <- diff$gene_name
# check
head(genelist,3)
# Aplnr Foxf1 Bmp5
# 13.45176 13.35322 12.02845
# load tpm
expr <- read.csv('tpm.csv')
# check
head(expr,3)
# gene_name day0_rep1 day0_rep2 day4_rep1 day4_rep2
# 1 Gnai3 155.689704 147.752705 361.38421 339.49937
# 2 Cdc45 68.347917 65.870974 61.79624 55.27016
# 3 H19 5.850431 7.558738 794.35331 637.19978Do enrichment:
# GO enrich
ego <- gseGO(geneList = genelist,
OrgDb = org.Mm.eg.db,
ont = "BP",
keyType = 'SYMBOL',
minGSSize = 100,
maxGSSize = 500,
pvalueCutoff = 0.05,
verbose = FALSE)Plot:
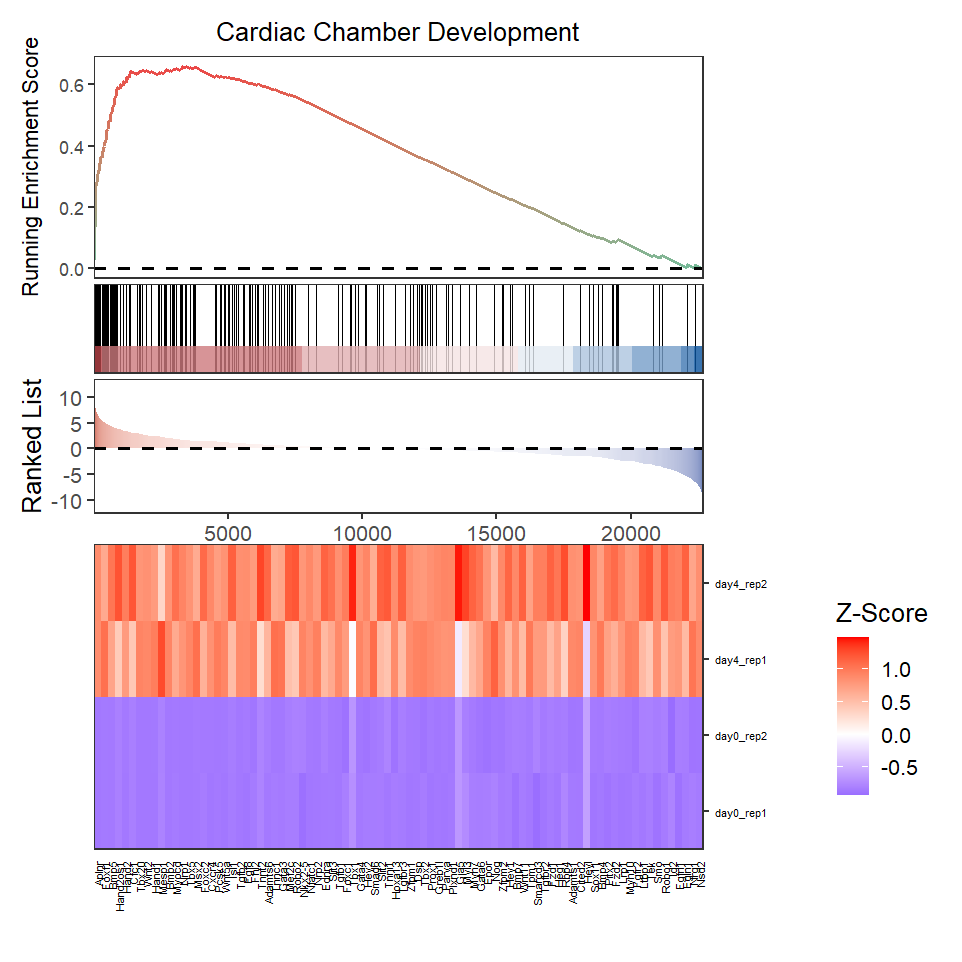
Remove gene rank plot:
# subplot
gseaNb(object = ego,
geneSetID = 'GO:0003205',
add.geneExpHt = T,
exp = expr,
subPlot = 2)
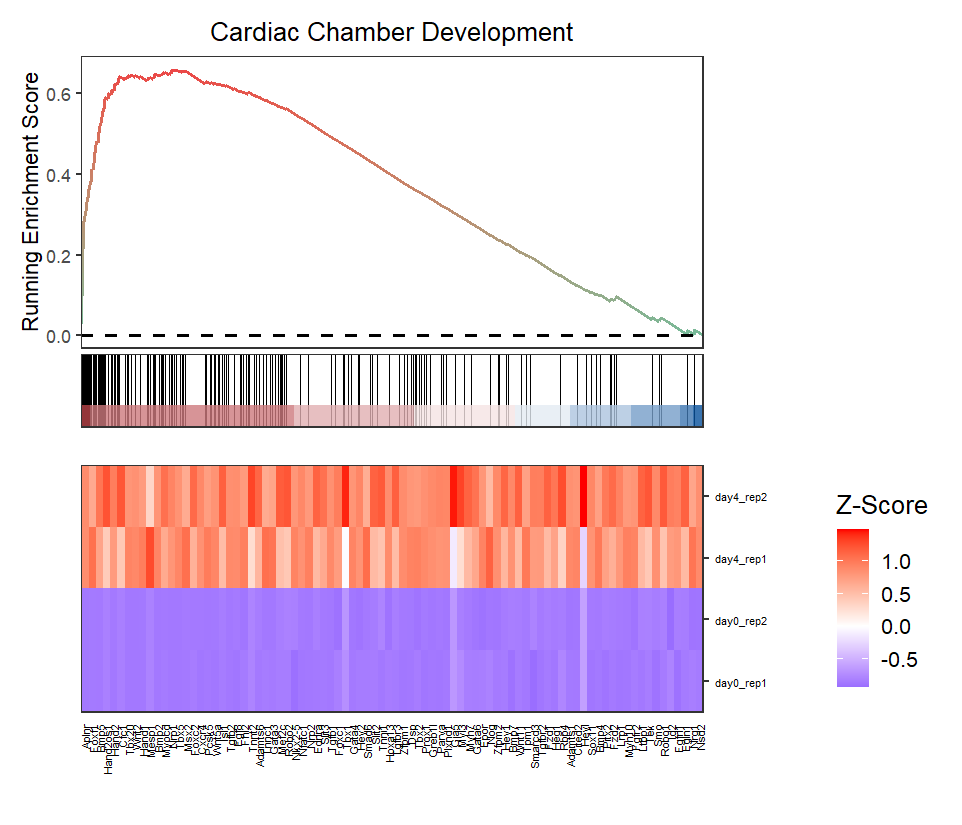
Change heatmap color:
# change heatmap colors
gseaNb(object = ego,
geneSetID = 'GO:0003205',
add.geneExpHt = T,
exp = expr,
subPlot = 2,
exp.col = c('green','black','red'))
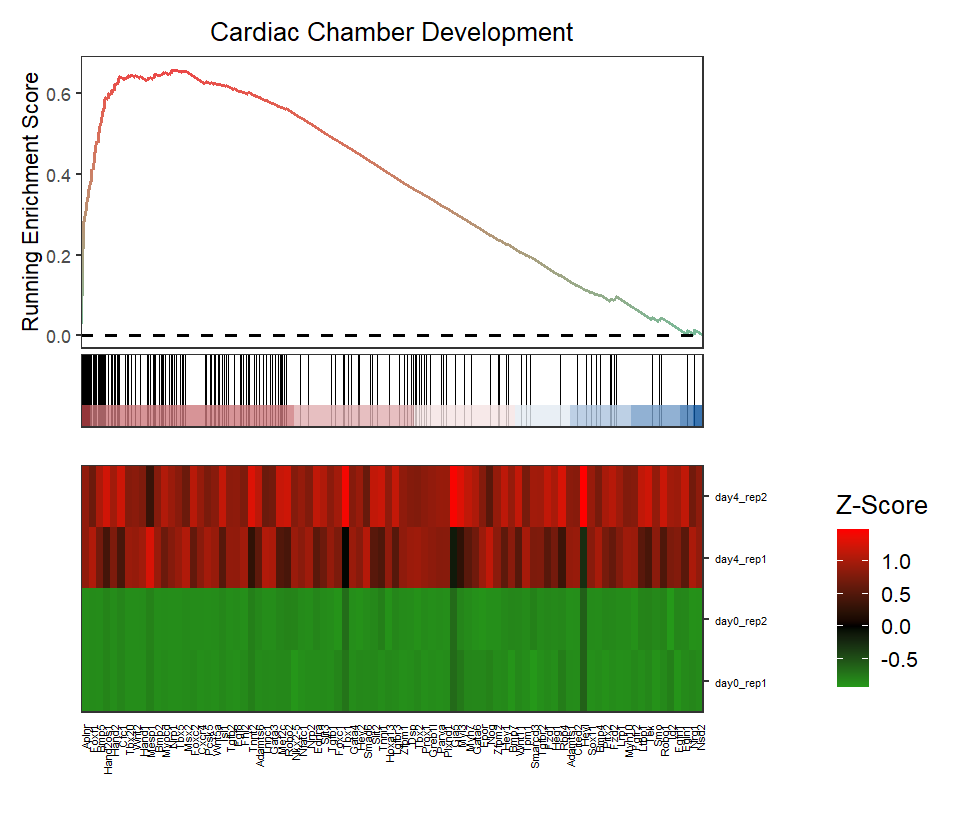
Change sample order:
# change heatmap sample orders
gseaNb(object = ego,
geneSetID = 'GO:0003205',
add.geneExpHt = T,
exp = expr,
subPlot = 2,
exp.col = c('green','black','red'),
sample.order = c(paste('day4_rep',1:2,sep = ''),
paste('day0_rep',1:2,sep = '')))
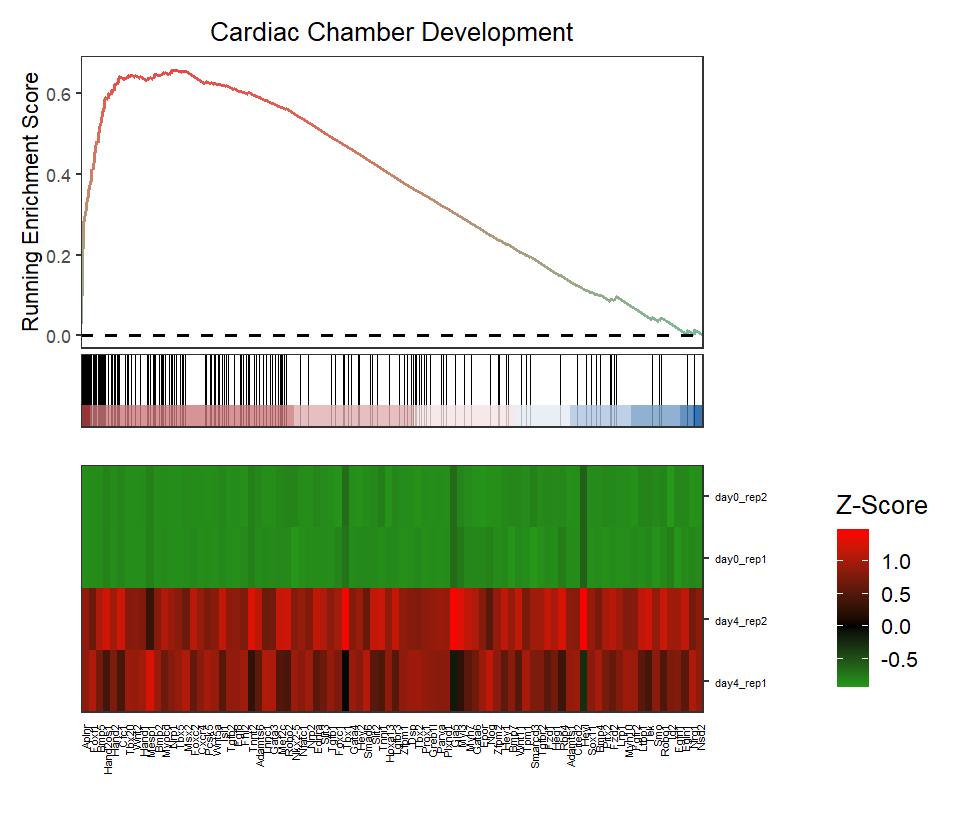
Change gene label size:
# change gene text size
gseaNb(object = ego,
geneSetID = 'GO:0007368',
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12)
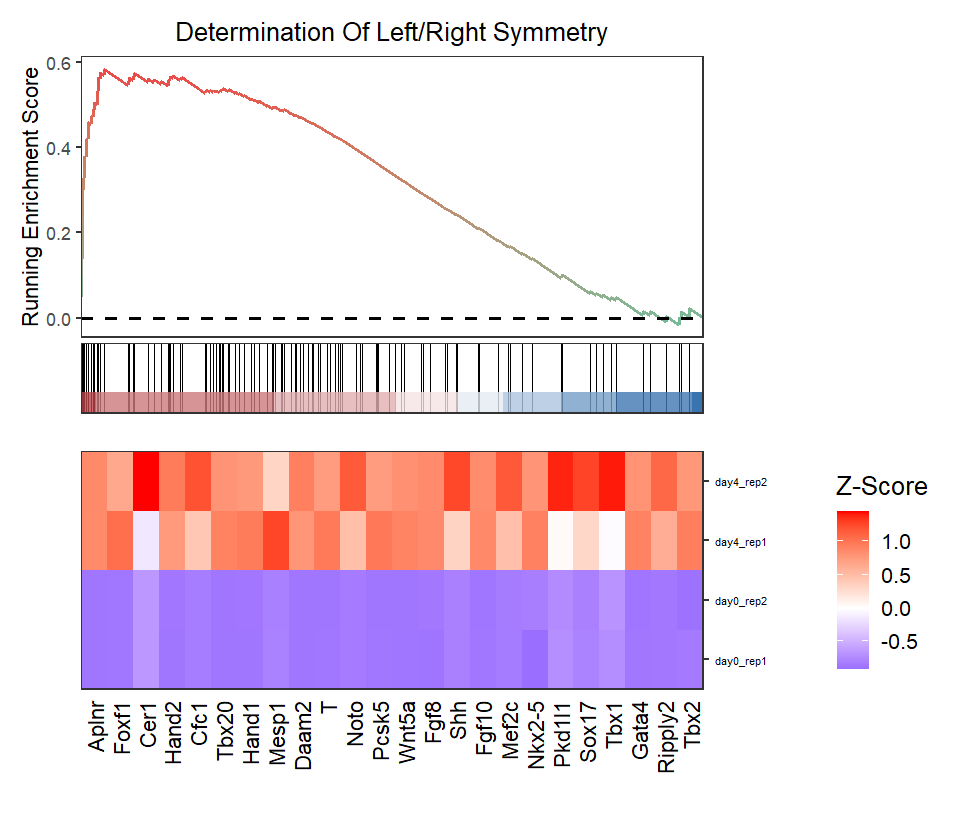
Turn off zscore:
# no scale expression
gseaNb(object = ego,
geneSetID = 'GO:0007368',
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12,
scale.exp = F)
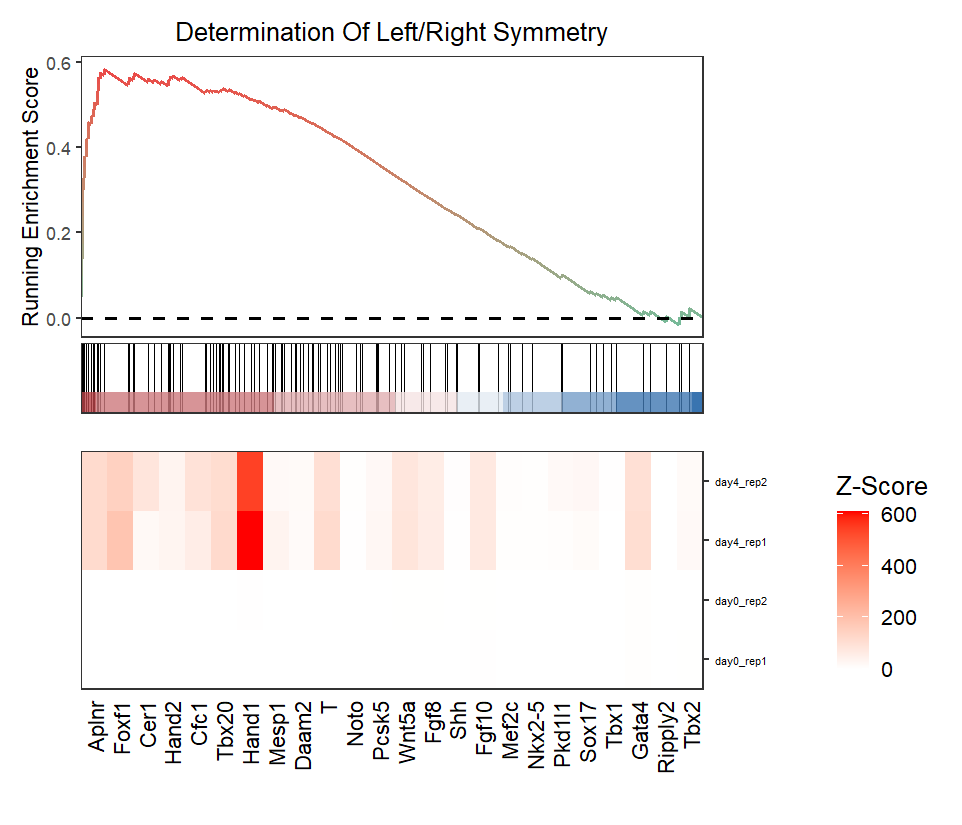
Control the relative height of heatmap:
# relative height
gseaNb(object = ego,
geneSetID = 'GO:0007368',
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12,
ght.relHight = 0.2)
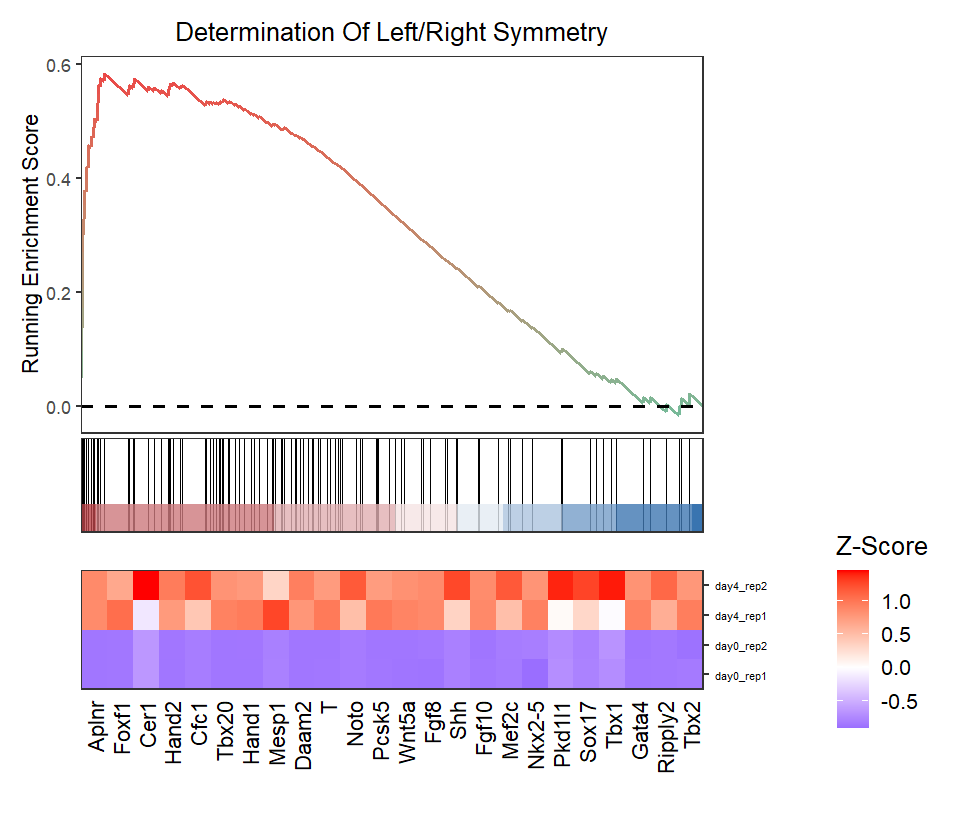
New style plot:
# new style gsea
gseaNb(object = ego,
geneSetID = 'GO:0007368',
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12,
newGsea = T)
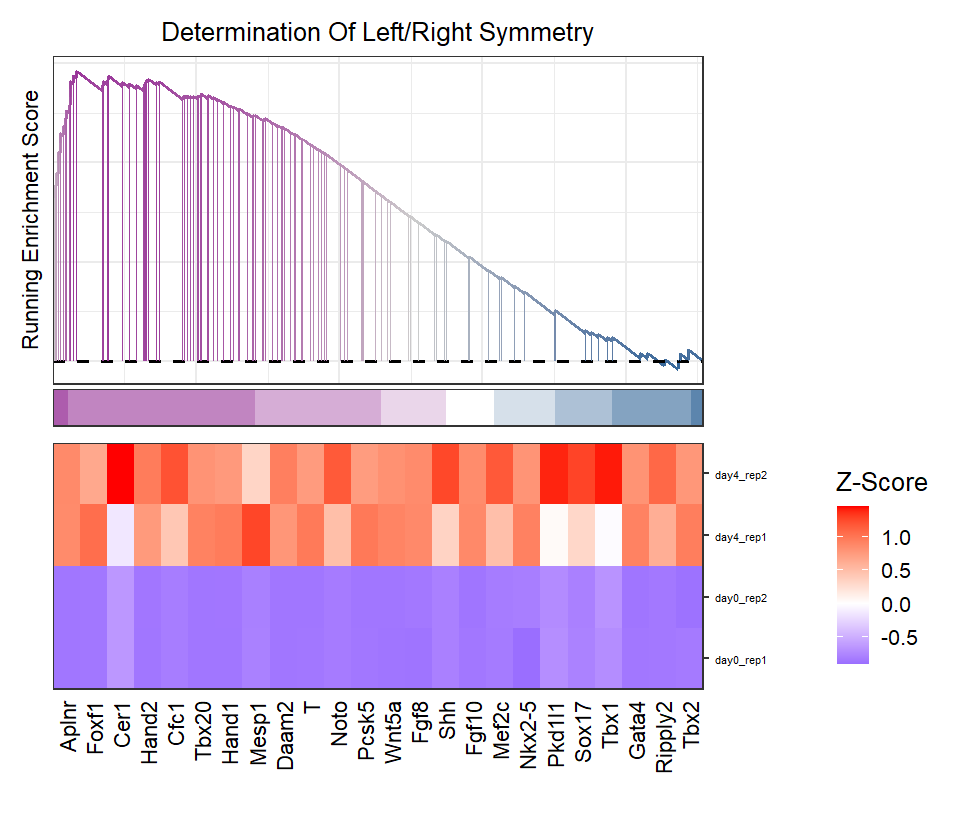
Multiple terms plot:
# multiple terms
gseaNb(object = ego,
geneSetID = c('GO:0003206','GO:0003007'),
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12,
addPval = T,
pvalX = 1,pvalY = 1)
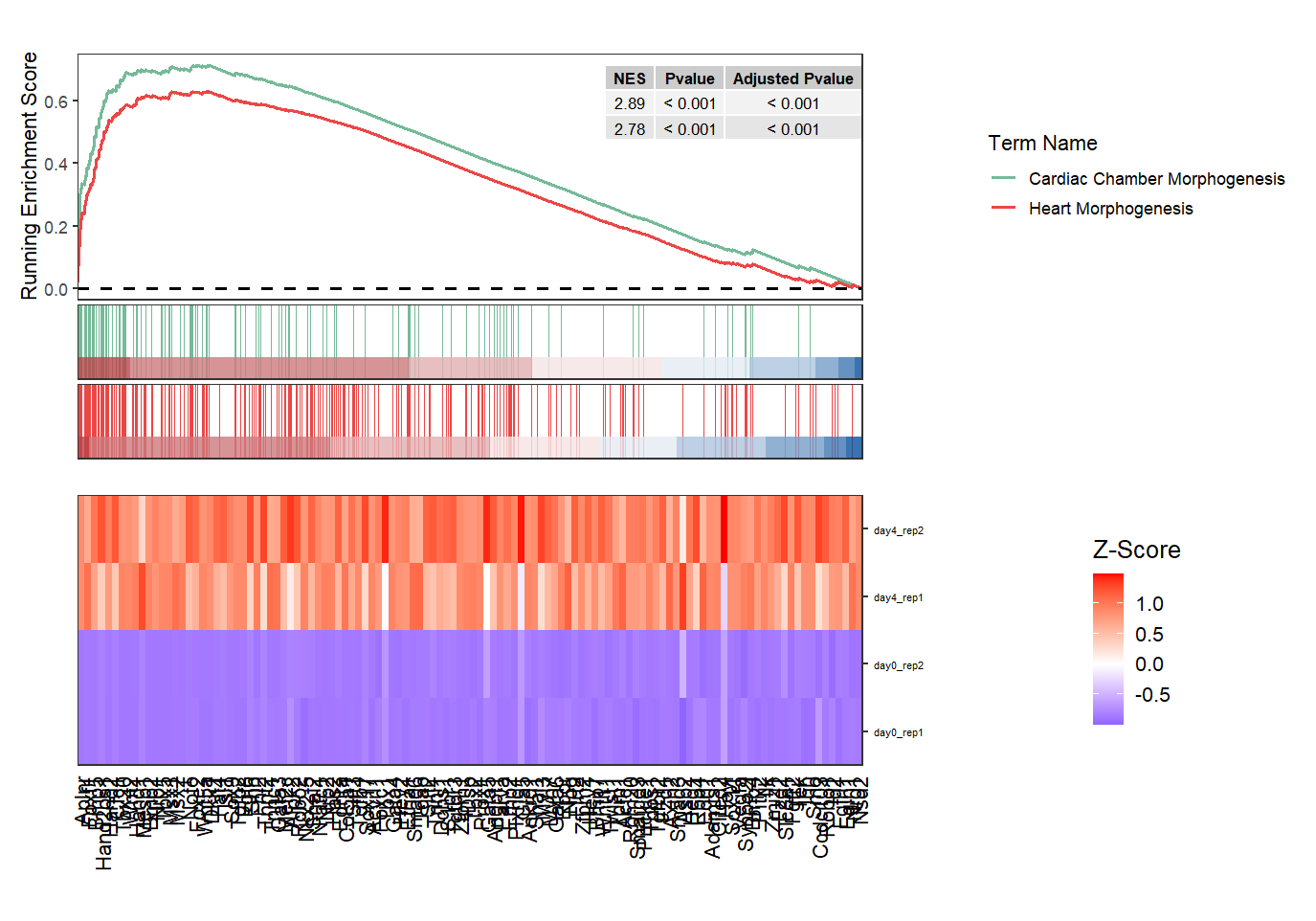
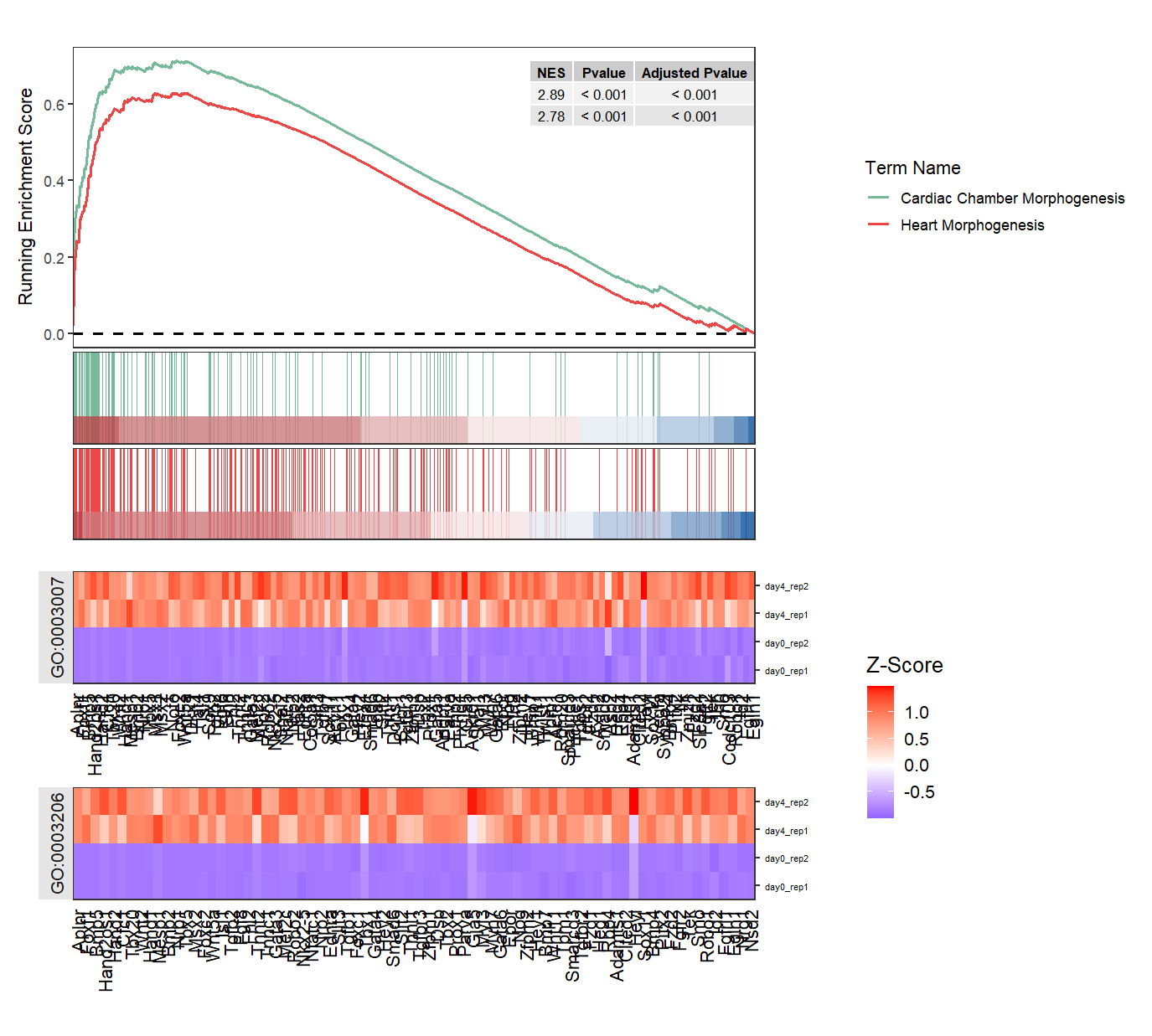
Make a facet for heatmap when multiple terms are shown:
# facet heatmap
gseaNb(object = ego,
geneSetID = c('GO:0007368','GO:0006304'),
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12,
addPval = T,
pvalX = 1,pvalY = 1,
ght.facet = T)
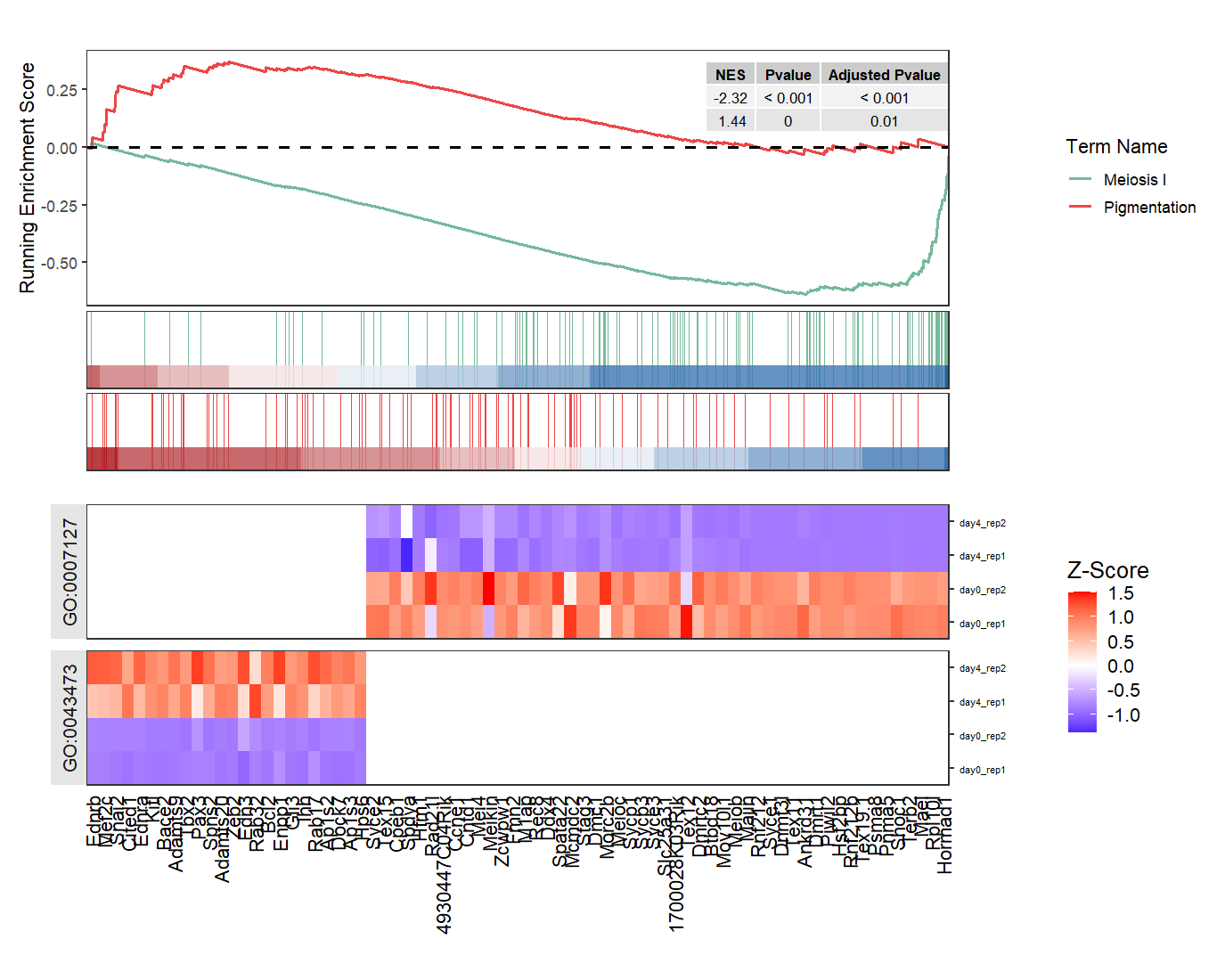
We select a up-regulated and down-regulated terms to show:
# adjust facet scales
gseaNb(object = ego,
geneSetID = c('GO:0007127','GO:0043473'),
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 12,
addPval = T,
pvalX = 1,pvalY = 1,
ght.facet = T,
ght.facet.scale = "fixed")
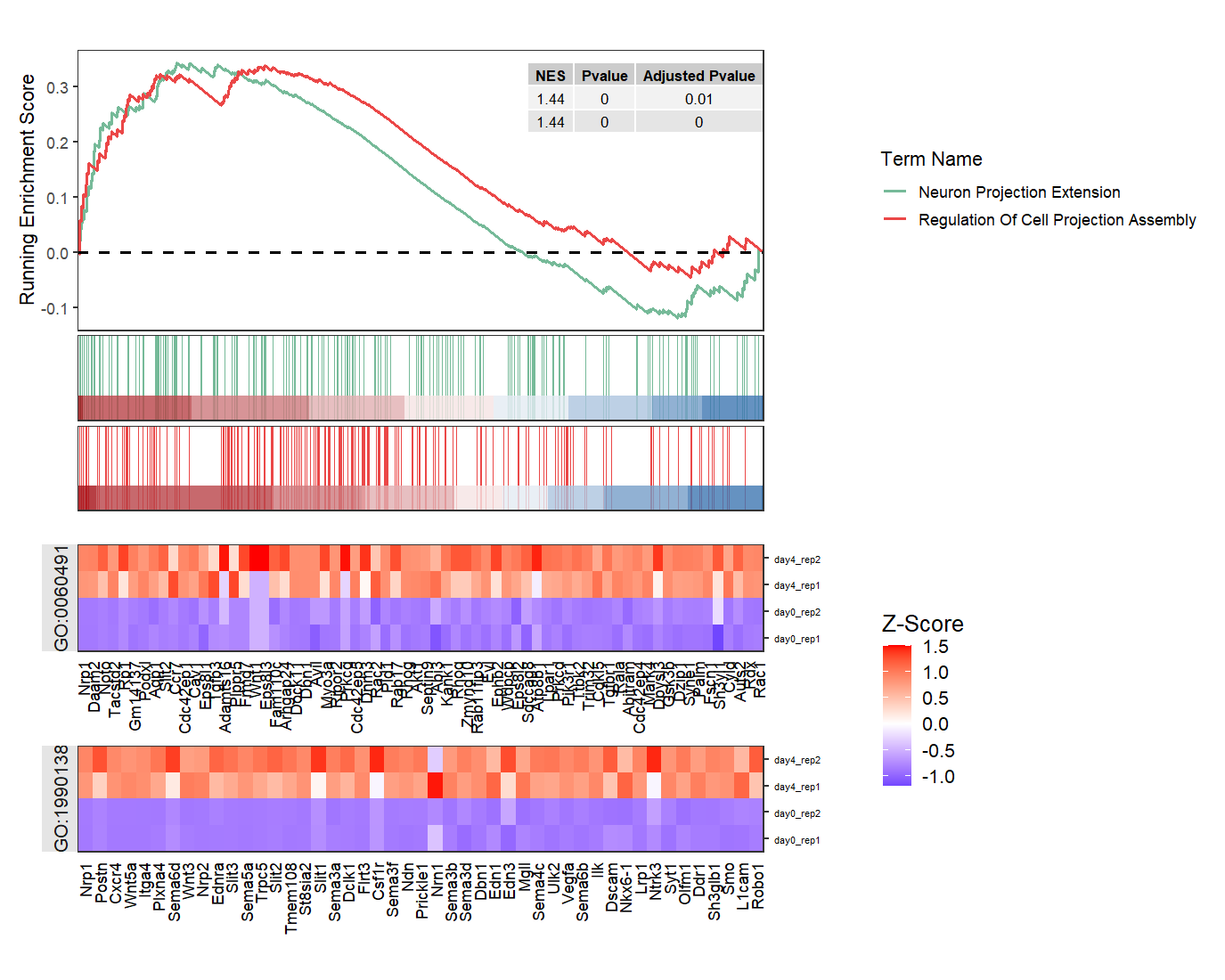
Some terms will share same genes:
# some terms share same genes
gseaNb(object = ego,
geneSetID = c('GO:1990138','GO:0060491'),
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 6,
addPval = T,
pvalX = 1,pvalY = 1,
ght.facet = T,
ght.facet.scale = "fixed")
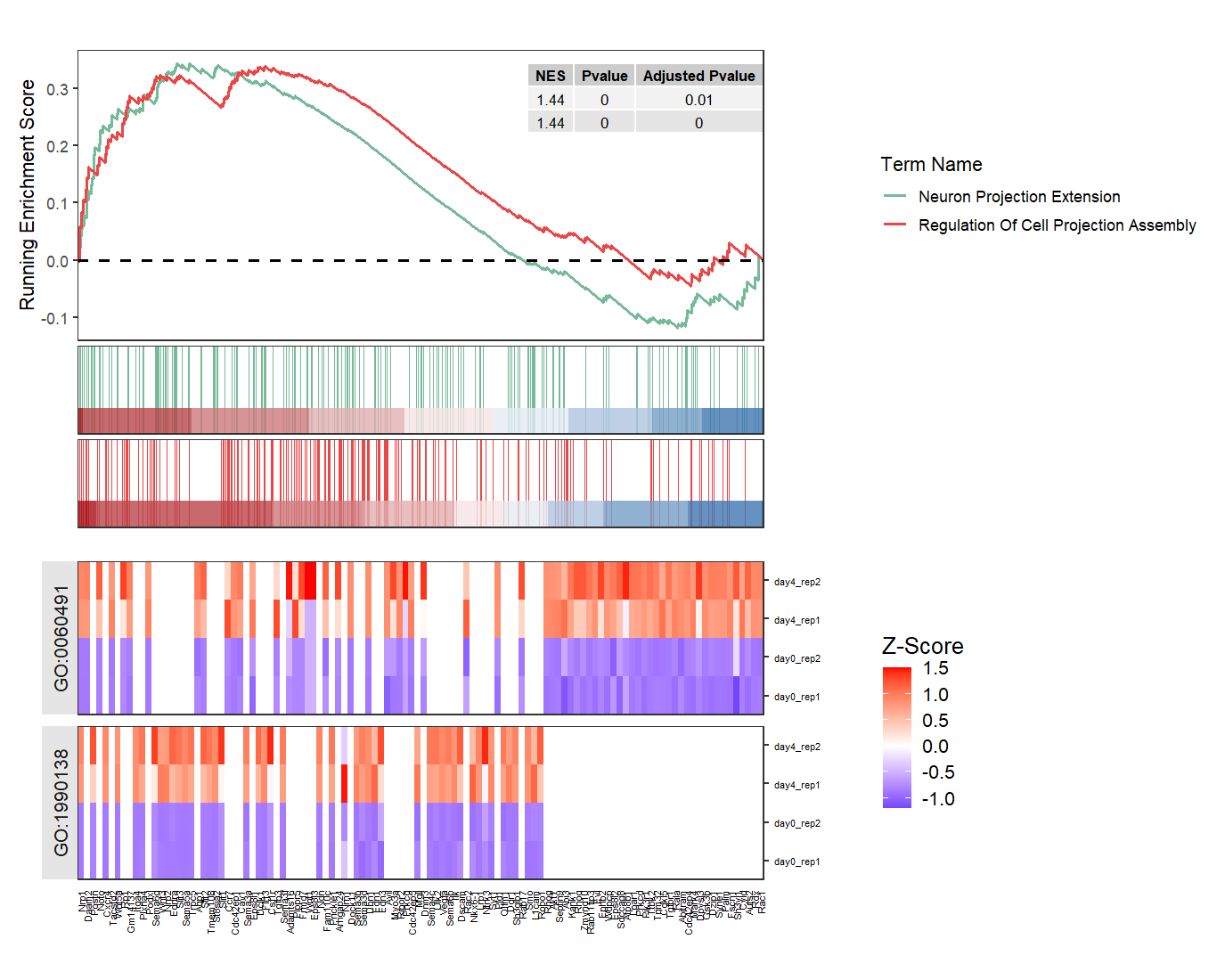
You can also set facet order:
# change facet order
gseaNb(object = ego,
geneSetID = c('GO:1990138','GO:0060491'),
add.geneExpHt = T,
exp = expr,
subPlot = 2,
ght.geneText.size = 10,
addPval = T,
pvalX = 1,pvalY = 1,
ght.facet = T,
termID.order = c('GO:0060491','GO:1990138'))