# Set the working directory to the alignment result folder
setwd("~/junjun_proj/31.ssb_proj/4.map-data/")
getwd()
# Install and load the riboTransVis package
devtools::install_github("junjunlab/riboTransVis", force = TRUE)
library(riboTransVis)
# Define input BAM files for total (translatome) and IP (interactome) libraries
tt.bam <- c("WT_Ssb1-GFP_translatome_rep1.bam",
"WT_Ssb1-GFP_translatome_rep2.bam",
"WT_Ssb2-GFP_translatome_rep1.bam",
"WT_Ssb2-GFP_translatome_rep2.bam")
ip.bam <- c("WT_Ssb1_interactome_rep1.bam",
"WT_Ssb1_interactome_rep2.bam",
"WT_Ssb2_interactome_rep1.bam",
"WT_Ssb2_interactome_rep2.bam")
# Construct the 'serp' object
obj <- construct_serp(genome_file = "../../index-data/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa",
gtf_file = "../../index-data/Saccharomyces_cerevisiae.R64-1-1.112.gtf",
mapping_type = "genome",
assignment_mode = "end5",
extend = TRUE,
extend_upstream = 50,
extend_downstream = 50,
total_bam_file = tt.bam,
total_sample_name = c("ssb1_rep1","ssb1_rep2","ssb2_rep1","ssb2_rep2"),
total_sample_group = c("ssb1","ssb1","ssb2","ssb2"),
IP_bam_file = ip.bam,
IP_sample_name = c("ssb1_rep1","ssb1_rep2","ssb2_rep1","ssb2_rep2"),
IP_sample_group = c("ssb1","ssb1","ssb2","ssb2"),
choose_longest_trans = TRUE)5 Serp object
Selective ribosome profiling (SeRP) is an advanced sequencing technique that builds upon traditional ribosome profiling to investigate translationally active ribosomes under specific conditions or bound to specific factors (e.g., chaperones, ligands, or drugs). Below, we outline its principles, methodology, and applications.
5.1 Principle
Selective ribosome profiling focuses on capturing ribosomes engaged in translating specific subsets of mRNAs or ribosomes interacting with regulatory molecules. Unlike standard ribosome profiling, which sequences all ribosome-protected mRNA fragments (RPFs), SeRP employs strategies to: 1. Enrich ribosomes of interest:
- Use translational inhibitors (e.g., harringtonine) to arrest ribosomes at initiation sites.
- Tag ribosomes (e.g., via affinity-tagged ribosomal proteins) for selective isolation.
- Target ribosomes bound to specific co-factors (e.g., chaperones like NAC or SRP).
2. Resolve context-specific translation: By isolating ribosomes in a defined state, SeRP reveals translational regulation in processes like stress responses, drug treatments, or pathogen-host interactions.
The general workflow is shown below Selective Ribosome Profiling to study interactions of translating ribosomes in yeast:
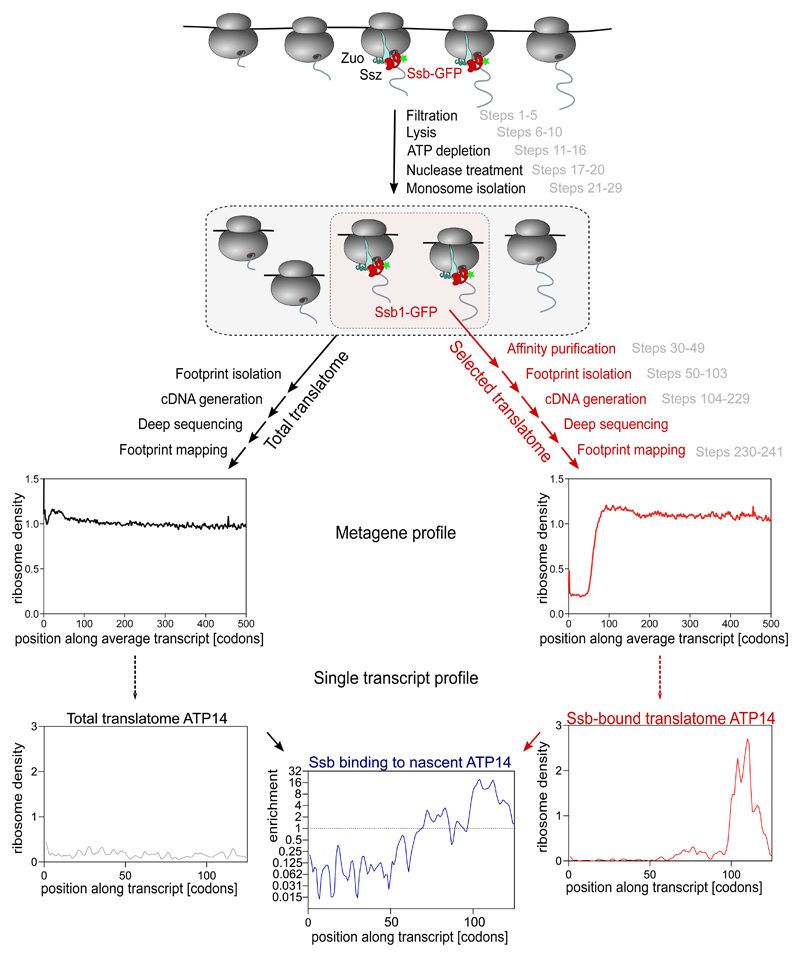
5.2 Methodology
The sequenced DNA libraries comprising the total translatome and the factor-bound translatome represent a rich data set to study factor engagement properties as well as features of mRNA translation. Initial steps of SeRP data analysis are generally performed using publicly available as well as customized read processing and alignment tools that are also used for RP analysis.
Initial read processing trims adaptors, removes low quality and noncoding reads, and aligns the filtered reads to the genome of interest. The riboTransVis can be used for subsequent analyses including ribosome E/P/A-site assignment, RP quality assessment and Basic SeRP analysis.
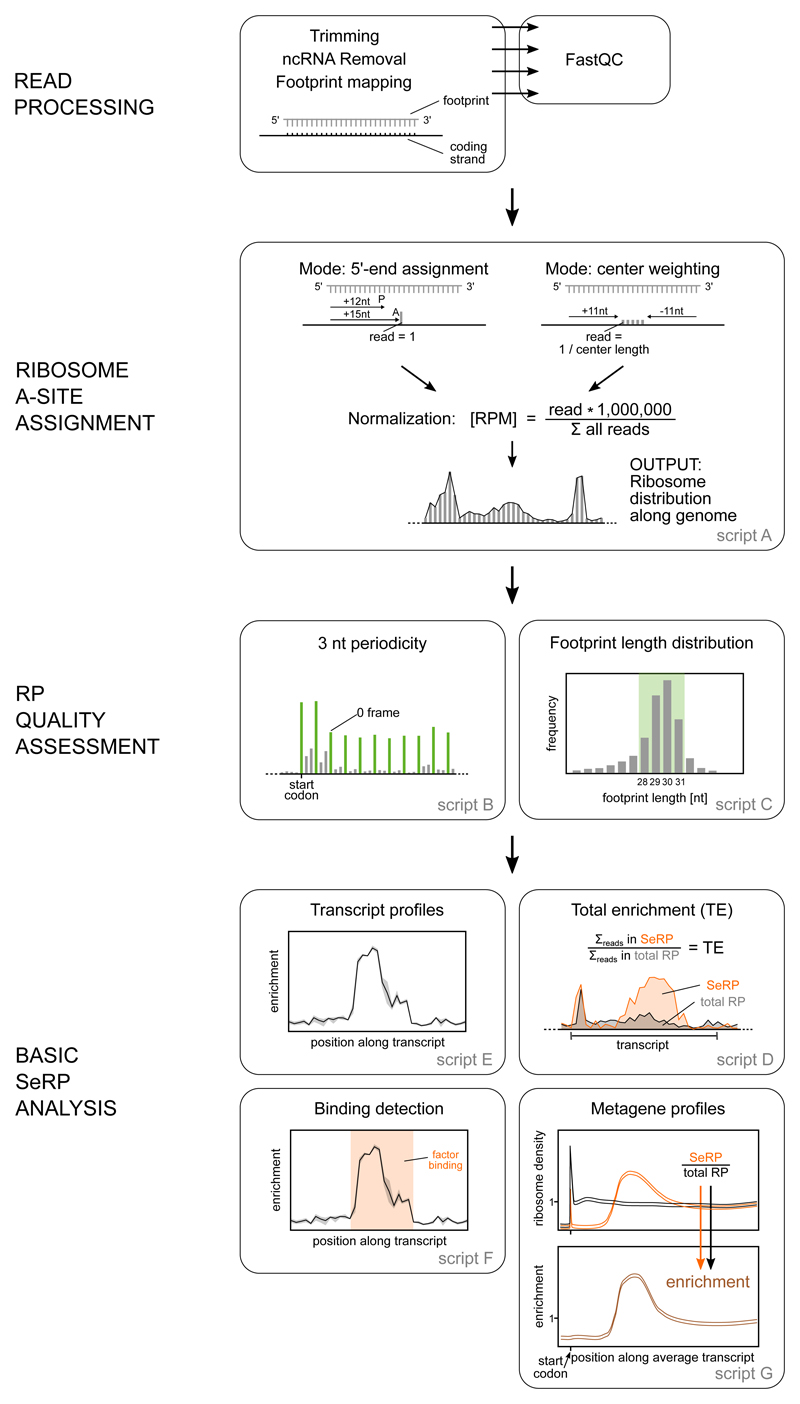
5.3 Serp object construction
We used the construct_serp() function from the riboTransVis package to create a serp object, which is designed for analyzing selective ribosome profiling data. This function takes as input both total and IP-enriched BAM files, along with annotation files and sample metadata.
5.4 Quality evaluation
We begin by evaluating basic quality control (QC) metrics, such as the distribution of read lengths—a key feature in ribosome profiling data, as it reflects the presence of ribosome-protected fragments of expected size:
# Compute summary statistics for total and IP libraries
obj <- generate_summary(
object = obj,
exp_type = c("total", "ip"),
nThreads = 40
)
# ==============================================================================
# qc
# Load ggplot2 for plotting
library(ggplot2)
length_plot(obj) +
scale_x_continuous(labels = scales::label_number(accuracy = 1)) +
facet_wrap(~sample, nrow = 2,scales = "free") +
theme_ribo()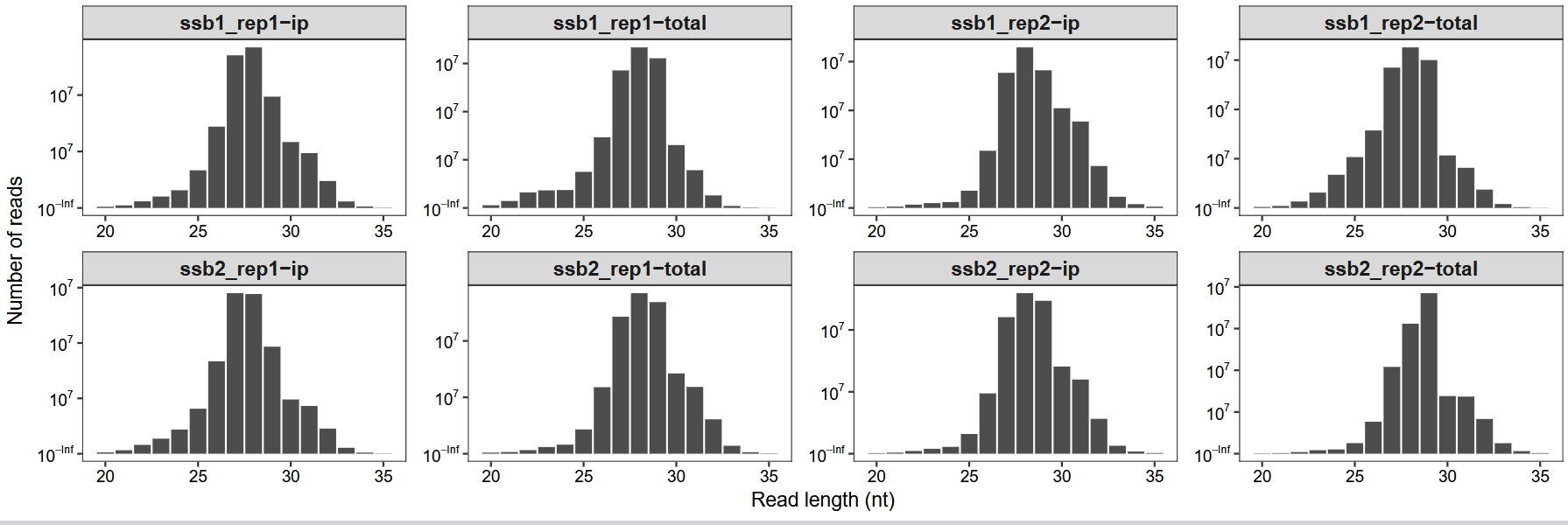
To assess translational periodicity, we examine the distribution of reads across three reading frames for different fragment lengths. This helps determine whether the data exhibit the expected 3-nt periodicity characteristic of actively translating ribosomes:
# Plot frame distribution across read lengths
length_plot(obj, type = "frame_length",
add_periodicity_label = FALSE) +
facet_wrap(~sample, nrow = 2, scales = "free") +
scale_fill_brewer(palette = "Greens", direction = -1) +
theme_ribo()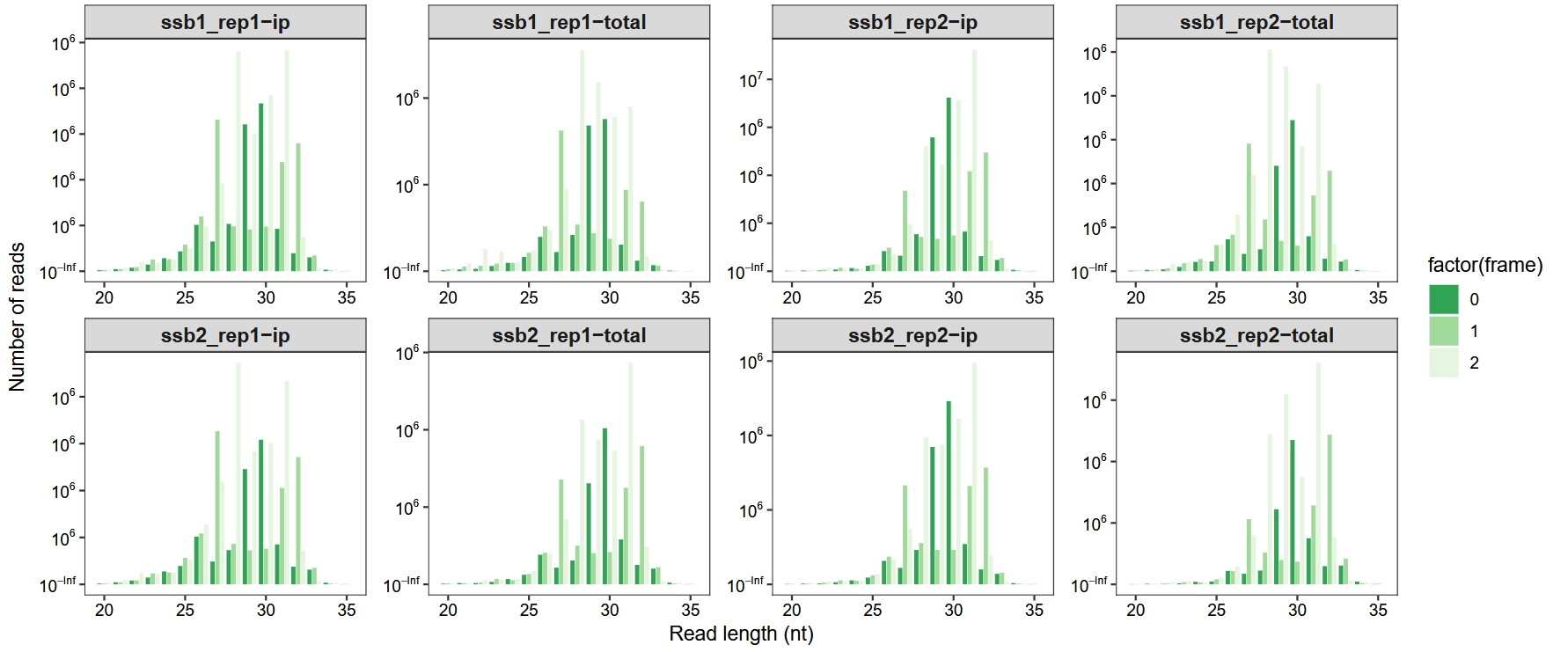
To further assess data quality and ribosome positioning accuracy, we examine the distribution of read offsets relative to annotated start codons. This helps verify whether reads of specific lengths show consistent positioning along transcripts:
# Plot relative offsets for reads of specific lengths
relative_offset_plot(obj, read_length = c(20, 35))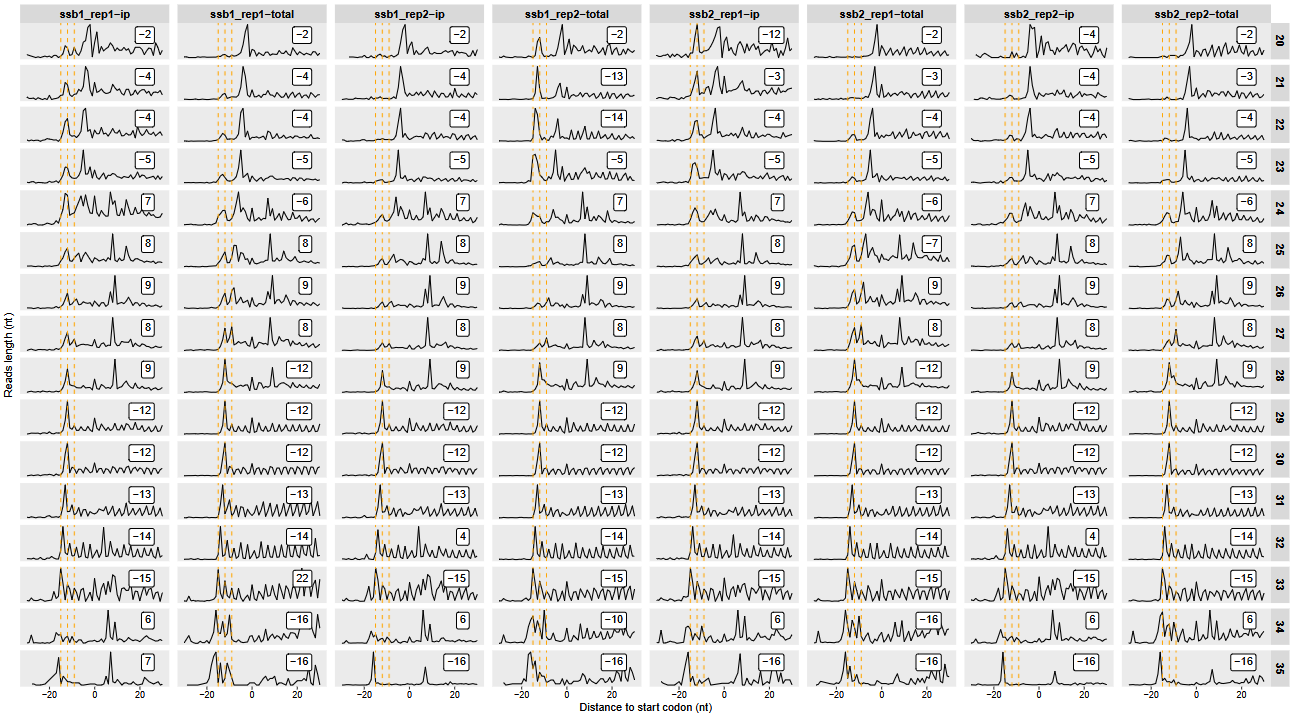
5.5 Offset correction
For accurate mapping of ribosome positions (P-sites), we manually assign offset values for reads of different fragment lengths. These offsets align the 5’ end of each read to the corresponding position within a ribosome, ensuring precise localization on transcripts:
# Define sample names
sp <- c("ssb1_rep1", "ssb1_rep2", "ssb2_rep1", "ssb2_rep2")
# Create a data frame specifying the offset (A-site position)
# for each read length (20–35 nt) and sample
offset <- data.frame(
sample = rep(c(paste(sp, "total", sep = "-"), paste(sp, "ip", sep = "-")), each = 16),
qwidth = rep(20:35, 8),
rel_pos = rep(c(rep(15, 11), rep(16, 5)), 8) # Set offset by read length
)
# Preview the offset table
head(offset)
# sample qwidth rel_pos
# 1 ssb1_rep1-total 20 15
# 2 ssb1_rep1-total 21 15
# 3 ssb1_rep1-total 22 15
# 4 ssb1_rep1-total 23 15
# 5 ssb1_rep1-total 24 15
# 6 ssb1_rep1-total 25 15
obj@reads_offset_info <- offset