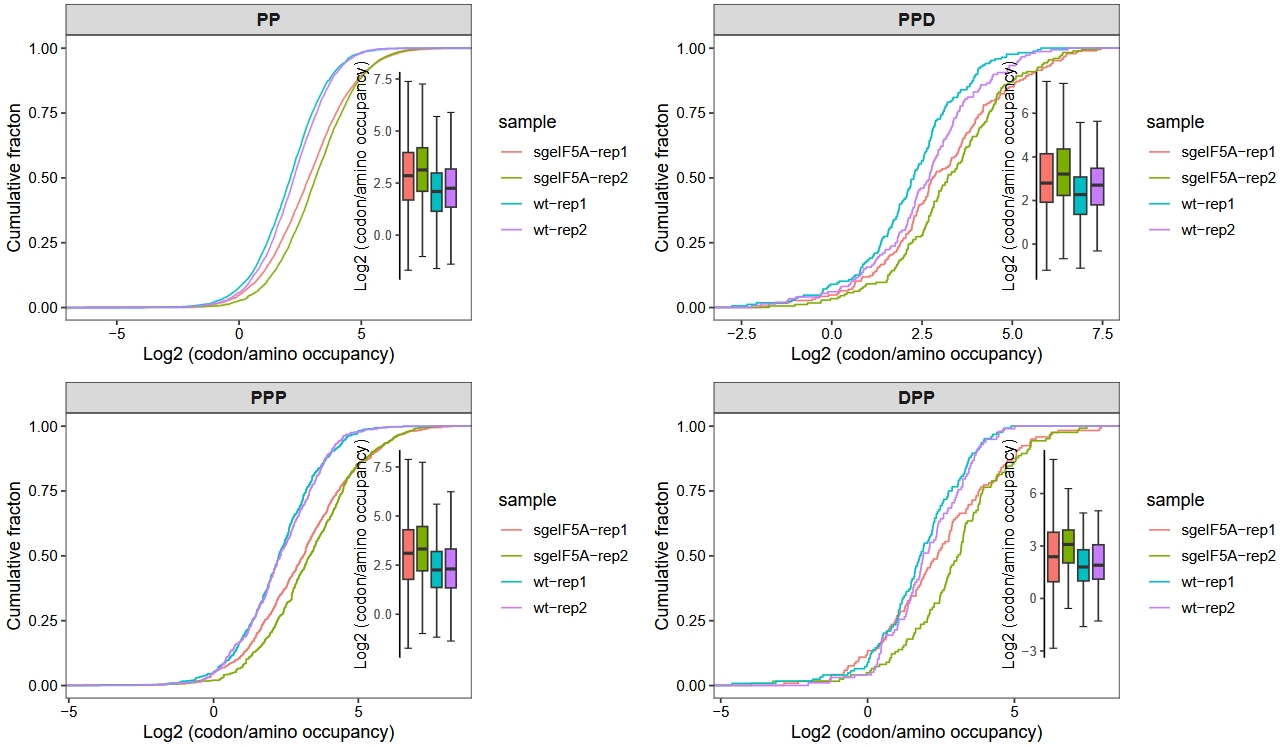
motif_occupancy(object = obj0,
cds_fa = "./sac_cds.fa",
search_type = "amino",
do_offset_correct = T,
motif_pattern = c("PP","PPP","PPD","DPP"))
# `summarise()` has grouped output by 'sample', 'rname'. You can override using the `.groups` argument.
# $plot
#
# $statistics
# # A tibble: 24 × 9
# codon_seq .y. group1 group2 p p.adj p.format p.signif method
# <chr> <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
# 1 PP value sgeIF5A-rep1 sgeIF5A-rep2 5.52e- 17 8.80e- 16 < 2e-16 **** Wilcoxon
# 2 PP value sgeIF5A-rep1 wt-rep1 5.45e-137 1.20e-135 < 2e-16 **** Wilcoxon
# 3 PP value sgeIF5A-rep1 wt-rep2 1.35e- 83 2.80e- 82 < 2e-16 **** Wilcoxon
# 4 PP value sgeIF5A-rep2 wt-rep1 1.13e-259 2.70e-258 < 2e-16 **** Wilcoxon
# 5 PP value sgeIF5A-rep2 wt-rep2 9.32e-180 2.10e-178 < 2e-16 **** Wilcoxon
# 6 PP value wt-rep1 wt-rep2 1.58e- 7 2.10e- 6 1.6e-07 **** Wilcoxon
# 7 PPD value sgeIF5A-rep1 sgeIF5A-rep2 1.13e- 1 4.5 e- 1 0.1129 ns Wilcoxon
# 8 PPD value sgeIF5A-rep1 wt-rep1 2.30e- 6 2.8 e- 5 2.3e-06 **** Wilcoxon
# 9 PPD value sgeIF5A-rep1 wt-rep2 1.69e- 2 1 e- 1 0.0169 * Wilcoxon
# 10 PPD value sgeIF5A-rep2 wt-rep1 3.35e- 11 5 e- 10 3.3e-11 **** Wilcoxon
# # ℹ 14 more rows
# # ℹ Use `print(n = ...)` to see more rowsMotif occupancy
Intro
To investigate how ribosome occupancy varies across different treatment conditions, cumulative distribution curves are employed to visualize the translational enrichment at specific amino acids or peptide motifs based on Ribo-seq data.
Cumulative curve
The motif_occupancy function allows for the quantification of ribosome occupancy at specific motifs or codons across individual transcripts, and generates cumulative distribution curves to illustrate global translational trends under different treatment conditions. As shown below, upon the loss of eIF5A, an overall increase in occupancy is observed at the following motifs:
By setting return_data = TRUE, the function returns the result as a data frame:
co <- motif_occupancy(object = obj0,
cds_fa = "./sac_cds.fa",
search_type = "amino",
do_offset_correct = T,
motif_pattern = c("PP","PPP","PPD","DPP"),
return_data = T)
# check
head(co)
# # A tibble: 6 × 7
# sample sample_group rname codon_pos value motif pos
# <chr> <chr> <chr> <dbl> <dbl> <chr> <int>
# 1 sgeIF5A-rep1 sgeIF5A YAL002W_mRNA|VPS8 47 34.0 PP 47
# 2 sgeIF5A-rep1 sgeIF5A YAL005C_mRNA|SSA1 462 5.69 PP 462
# 3 sgeIF5A-rep1 sgeIF5A YAL011W_mRNA|SWC3 339 10.9 PP 339
# 4 sgeIF5A-rep1 sgeIF5A YAL013W_mRNA|DEP1 164 13.8 PP 164
# 5 sgeIF5A-rep1 sgeIF5A YAL014C_mRNA|SYN8 149 12.0 PP 149
# 6 sgeIF5A-rep1 sgeIF5A YAL015C_mRNA|NTG1 327 3.22 PP 327Visualize the cumulative distribution curve of ribosome occupancy across codons:
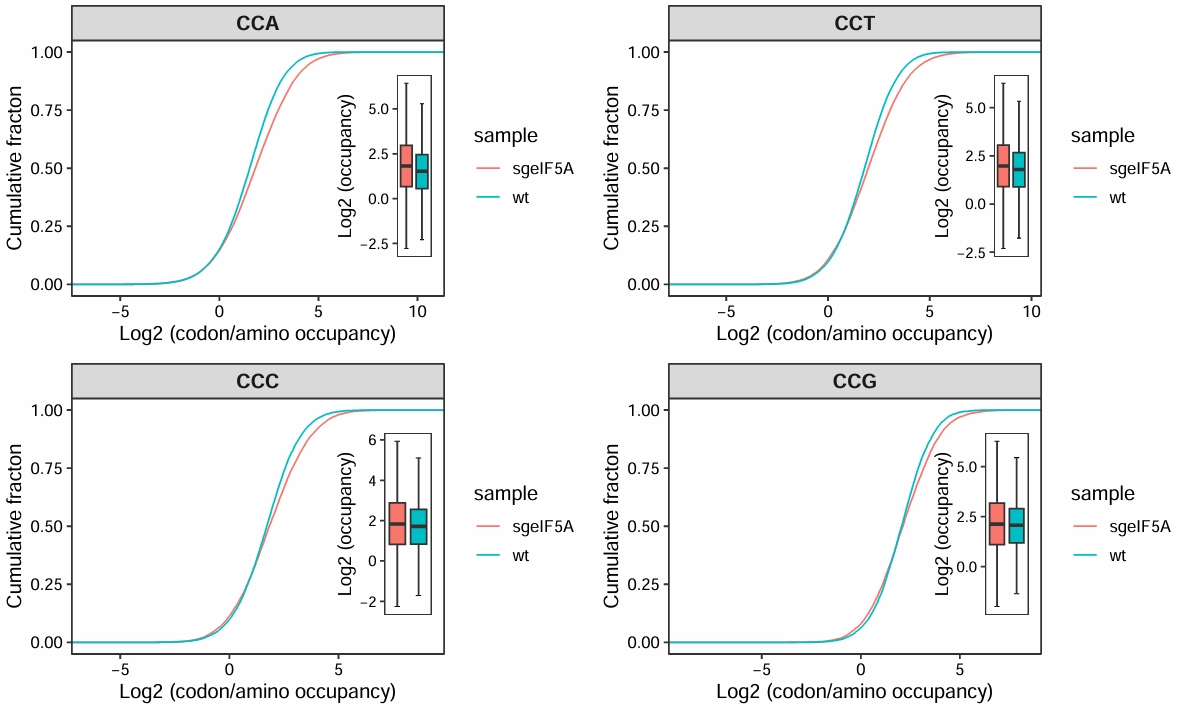
motif_occupancy(object = obj0,
cds_fa = "./sac_cds.fa",
search_type = "codon",
do_offset_correct = T,
motif_pattern = c("CCA","CCT","CCG","CCC"))
# `summarise()` has grouped output by 'sample', 'rname'. You can override using the `.groups` argument.
# Adding missing grouping variables: `sample_group`, `freq`
# $plot
#
# $statistics
# # A tibble: 24 × 9
# codon_seq .y. group1 group2 p p.adj p.format p.signif method
# <chr> <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
# 1 CCA value sgeIF5A-rep1 sgeIF5A-rep2 2.36e-107 4.30e-106 < 2e-16 **** Wilcoxon
# 2 CCA value sgeIF5A-rep1 wt-rep1 2.32e-141 4.6 e-140 < 2e-16 **** Wilcoxon
# 3 CCA value sgeIF5A-rep1 wt-rep2 3.61e- 9 1.4 e- 8 3.6e-09 **** Wilcoxon
# 4 CCA value sgeIF5A-rep2 wt-rep1 0 0 < 2e-16 **** Wilcoxon
# 5 CCA value sgeIF5A-rep2 wt-rep2 2.80e-191 6.20e-190 < 2e-16 **** Wilcoxon
# 6 CCA value wt-rep1 wt-rep2 7.57e- 96 1.20e- 94 < 2e-16 **** Wilcoxon
# 7 CCT value sgeIF5A-rep1 sgeIF5A-rep2 5.02e- 99 8.50e- 98 < 2e-16 **** Wilcoxon
# 8 CCT value sgeIF5A-rep1 wt-rep1 4.70e- 62 5.6 e- 61 < 2e-16 **** Wilcoxon
# 9 CCT value sgeIF5A-rep1 wt-rep2 8.34e- 5 8.3 e- 5 8.3e-05 **** Wilcoxon
# 10 CCT value sgeIF5A-rep2 wt-rep1 0 0 < 2e-16 **** Wilcoxon
# # ℹ 14 more rows
# # ℹ Use `print(n = ...)` to see more rows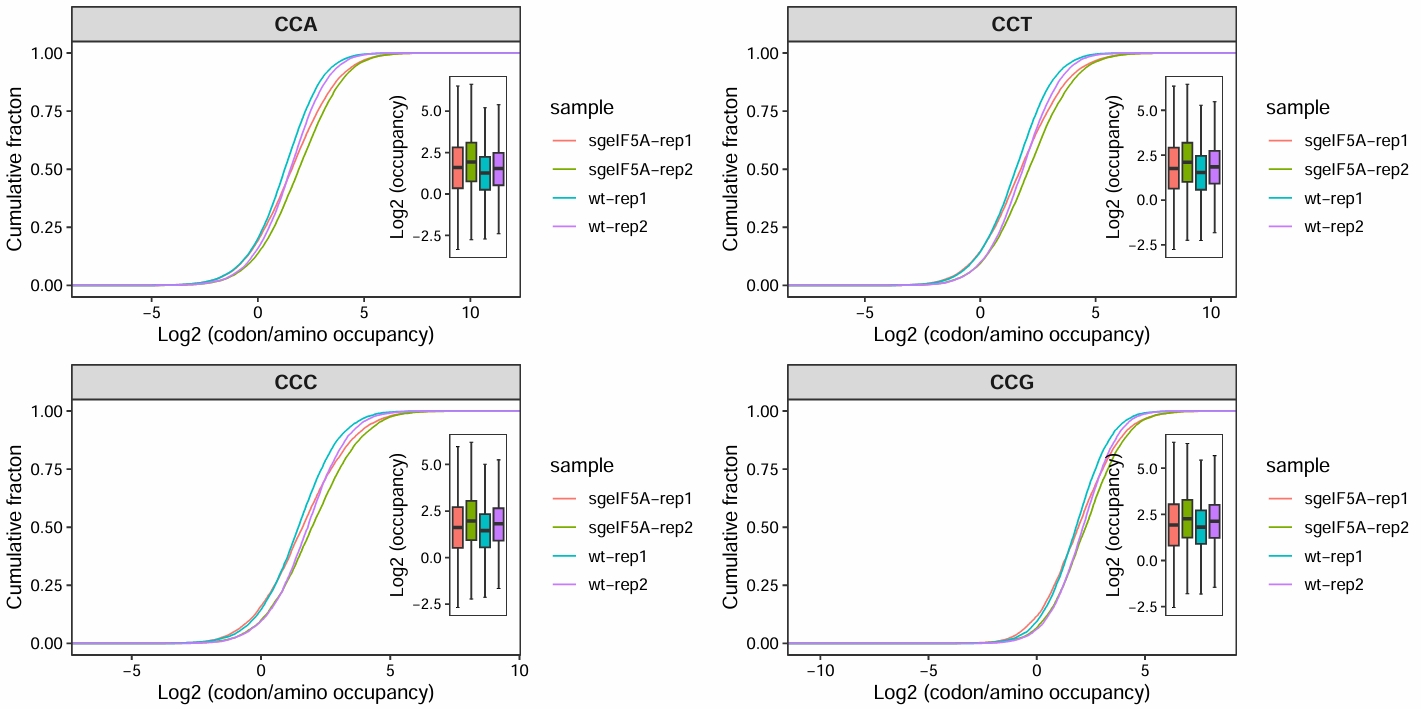
Merge replicates:
motif_occupancy(object = obj0,
cds_fa = "./sac_cds.fa",
search_type = "codon",
do_offset_correct = T,
merge_rep = T,
motif_pattern = c("CCA","CCT","CCG","CCC"))
# `summarise()` has grouped output by 'sample', 'rname'. You can override using the `.groups` argument.
# $plot
#
# $statistics
# # A tibble: 4 × 9
# codon_seq .y. group1 group2 p p.adj p.format p.signif method
# <chr> <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
# 1 CCA value sgeIF5A wt 1.85e-142 7.4 e-142 < 2e-16 **** Wilcoxon
# 2 CCT value sgeIF5A wt 3.55e- 53 1.10e- 52 < 2e-16 **** Wilcoxon
# 3 CCC value sgeIF5A wt 1.76e- 15 3.5 e- 15 1.8e-15 **** Wilcoxon
# 4 CCG value sgeIF5A wt 9.70e- 5 9.70e- 5 9.7e-05 **** Wilcoxon